sclerosi multipla
sclerosi multipla
Malattia infiammatoria demielinizzante cronica a carico del sistema nervoso centrale.
Epidemiologia
La s. m. è più frequente nel sesso femminile (2:1), con esordio in genere fra i 20 e i 40 anni, anche se non sono rari i casi a esordio nella età pediatrica o nella età avanzata. Numerosi studi hanno evidenziato un rapporto fra prevalenza di malattia e latitudine, con una maggiore frequenza con l’aumentare della latitudine, sia nell’emisfero settentrionale sia in quello meridionale. Nell’Europa settentrionale la prevalenza è di circa 120÷200 casi ogni 100.000 abitanti e in Italia circa 100 persone su 100.000 ne sono affette; in partic., in Sardegna la prevalenza della malattia arriva a 150 casi su 100.000. L’incidenza della malattia, ossia il numero di nuovi casi/anno, è di ca. 3÷5 casi ogni 100.000 persone.
Eziopatogenesi
La s. m. è ritenuta una malattia autoimmune organo-specifica, sulla base di evidenze cliniche e sperimentali. In partic., è possibile riprodurre nell’animale una malattia molto simile, l’encefalite autoimmune sperimentale (EAE), iniettando omogenati di mielina, proteine mieliniche o frazioni di proteine, anche non mieliniche, e la malattia può essere trasferita passivamente utilizzando cellule linfocitarie T da un animale malato a uno singenico sano. Tutti gli studi concordano sull’esistenza di una suscettibilità geneticamente determinata a contrarre la malattia, su cui pur agiscono fattori ambientali. Mentre il rischio di ammalarsi di s. m. è, come si è detto, di ca. 1 su 1.000 nella popolazione generale, la probabilità di sviluppare una s. m. aumenta all’1÷3% nei parenti di primo grado di una persona affetta. La regione genomica più importante è il complesso maggiore di istocompatibilità, situato sul cromosoma 6, in partic. l’allele Hladr2 o, in Sardegna, l’Hladr4, ma esistono numerosi geni, tutti coinvolti con le funzioni immuni e tutti di modesta dimensione, che conferiscono un aumentato rischio a contrarre la malattia. I fattori ambientali legati alla malattia, non ancora noti, agirebbero principalmente nei primi 15 anni di vita, perché studi di migrazione mostrano che i migranti conservano il rischio della zona di origine quando la migrazione avviene dopo i 15 anni o acquisiscono il rischio del nuovo Paese quando la migrazione avviene prima. La malattia è causata da una attivazione di linfociti T autoreattivi che passano la barriera ematoencefalica e colpiscono antigeni mielinici o non mielinici del sistema nervoso. Successivamente entrano nell’SNC anche linfomonociti, non specificamente attivati, che insieme ad anticorpi, citochine proinfiammatorie e chemochine danneggiano il tessuto nervoso. Le lesioni si manifestano attraverso placche di demielinizzazione, cui si associa un importante danno assonale. Qualunque area dell’SNC può essere affetta, ma costante è l’interessamento delle zone periventricolari, intorno al IV ventricolo e all’acquedotto, del corpo calloso, dei nervi ottici, del tronco encefalico, delle aree sottocorticali e del midollo spinale. La malattia non è limitata alle aree o placche di demielinizzazione, ma interessa, anche se in misura più lieve e diffusamente, la sostanza bianca apparentemente normale, ove sono presenti focolai di attivazione microgliale e di danno assonale, e la sostanza grigia, sia corticale sia sottocorticale.
Clinica
Può essere interessata dalla patologia qualunque funzione del sistema nervoso, anche se alcuni sintomi sono più frequenti di altri. I disturbi più comuni nella malattia sono: diminuzione della forza a uno o più arti, con le caratteristiche della paralisi centrale piramidale e associata ipertonia spastica e disturbi della sensibilità, con parestesie (abnorme sensazione di formicolio, punture di spillo, bruciore, in assenza di una stimolazione esterna), o una riduzione della sensibilità (ipoestesia) o, al contrario, un aumento della sensibilità (iperestesia) o una trasformazione dello stimolo (disestesia). Non rari i dolori, talvolta di tipo neuropatico (dolori folgoranti, fastidiosi, subcontinui o parossistici) o di tipo muscoloscheletrico, da malposizioni, specie lombosacrali, e disturbi cerebellari, che si esprimono con alterazioni della coordinazione del movimento, tremore agli arti, atassia della marcia. La neurite ottica, molto frequente, è spesso un sintomo di esordio che si caratterizza per un importante calo della acuità visiva con annebbiamento della vista non correggibile con lenti; possono presentarsi diplopia per paralisi dei nervi oculomotori, turbe sfinteriche, con minzione imperiosa, ritenzione urinaria, stipsi, turbe sessuali, con difficoltà di erezione e calo della libido. Non rare sono le problematiche depressive, più spesso nelle fasi iniziali della malattia, e i disturbi cognitivi, con problemi di memoria, difficoltà attentive, che raramente sono di entità tale da configurare un quadro di grave decadimento mentale. La fatica è un sintomo molto frequente, e non rare sono le vertigini, l’ipoacusia, in genere unilaterale, la cefalea. I disturbi neurologici e la conseguente disabilità sono quantificati in base a scale di valutazione, di cui la più utilizzata è quella di Kurtzke (EDSS, Expanded Disability Status Scale).
Diagnosi, decorso e prognosi
La diagnosi di s. m. si basa sulla presenza documentata di disseminazione temporale, con almeno due episodi separati nel tempo di sofferenza focale dell’SNC, e di disseminazione spaziale, con la compromissione come minimo di due diverse aree dell’SNC. La disseminazione spaziale o temporale della malattia può essere anche dimostrata da esami paraclinici, come la RM o i potenziali evocati. Il decorso della malattia può essere molto variabile e la prognosi nel singolo caso difficile. Tuttavia, nella maggior parte dei casi, il decorso è relativamente stereotipato: l’esordio è intorno ai 20÷40 anni, l’evoluzione è a ricadute (decorso a ricadute e remissioni), con iniziale recupero del deficit lamentato, anche completo, e comparsa, dopo alcuni anni, di esiti permanenti; dopo 10÷20 anni dall’esordio si rende evidente un decorso progressivo (decorso secondariamente progressivo), con importanti problemi di deambulazione e di autonomia che possono portare all’uso della carrozzina intorno ai 50÷60 anni di età. In ca. il 15% dei casi la malattia è progressiva fin dall’inizio (decorso primariamente progressivo), con o senza ricadute. Nel 15÷20% dei casi il decorso è benigno, con modesta disabilità dopo 15÷20 anni dall’esordio e circa il 4÷5% dei casi ha un decorso ‘maligno’, con importante disabilità entro pochi anni dall’esordio. La prognosi può essere difficile nel caso singolo, anche se frequenti ricadute nei primi anni di malattia, la precoce comparsa di segni piramidali o cerebellari, un decorso progressivo e un importante carico lesionale alla RM hanno un significato prognostico negativo.
Esami diagnostici complementari
Per la diagnosi sono di cruciale importanza:
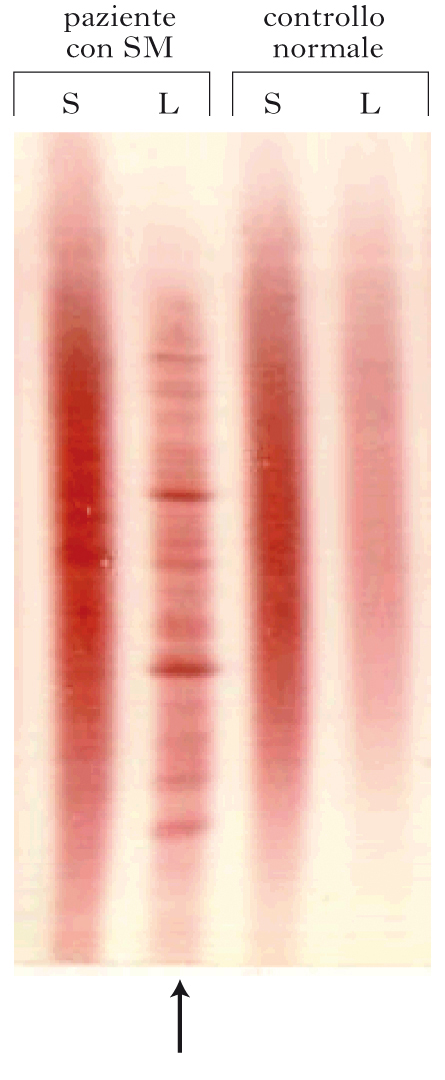
• l’analisi del liquor cefalorachidiano, che mostra un aumento liquorale delle immunoglobuline G (IgG), con all’immunoblotting la presenza di bande oligoclonali in ca. il 95% dei casi;
• i potenziali evocati, visivi, somatosensoriali o motori, in grado di evidenziare una compromissione delle vie nervose, anche asintomatica;
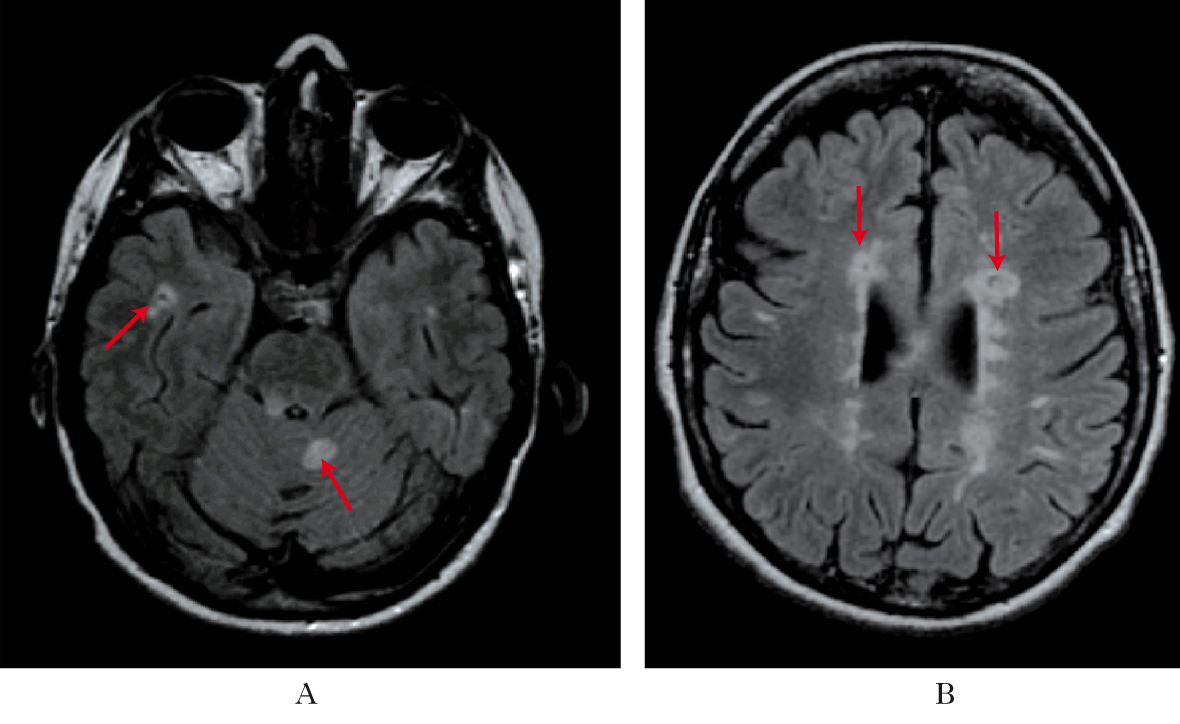
• la risonanza magnetica, per evidenziare aree iperintense nella sostanza bianca degli emisferi cerebrali, spesso disposte a sede periventricolare, iuxtacorticale e sottotentoriale, e, in caso di attività, aree che prendono contrasto; con il progredire della malattia, nelle fasi avanzate, possono diventare evidenti segni di atrofia cerebrale diffusa. La RM mostra che la malattia è attiva anche quando sembra clinicamente silente, e solo una su circa 10 lesioni che si evidenziano alla RM ha espressività clinica.
Terapia
Le cure a disposizione sono relativamente efficaci nelle fasi iniziali di malattia, quando predomina l’infiammazione, mentre sono di utilità molto modesta quando è iniziata la fase progressiva della malattia, ove predomina la degenerazione assonale. La terapia delle ricadute consiste nell’uso di steroidi, generalmente utilizzati ad alto dosaggio per pochi giorni e per via endovenosa (metilprednisolone) o, meno frequentemente, anche per via orale o intramuscolare. La terapia immunomodulante con interferone b1b o interferone b1a o glatiramer acetato, per via sottocutanea o intramuscolare, a diverse posologie, riduce la frequenza delle ricadute del 32÷34% e rallenta la progressione della disabilità, mentre nelle forme secondariamente progressive solo l’interferone b1b è stato dimostrato efficace nel rallentare, in misura modesta, la disabilità, specie nei casi con associate ricadute e che hanno iniziato la fase progressiva da non molto tempo. Il natalizumab è un anticorpo monoclonale che riduce di circa il 68% la frequenza delle ricadute e del 42% la progressione della disabilità, ma ha il rischio (circa 1 su 1.000, specie dopo il secondo anno di terapia) di causare lo sviluppo di una leucoencefalopatia multifocale progressiva (PML, Progressive Multifocal Leukoencephalopathy), una grave encefalite virale. fra le terapie immunosoppressive approvate in Italia, il mitoxantrone ha una cardiotossicità cumulativa e un rischio di leucemia non trascurabile; frequentemente utilizzati sono l’azatioprina e la ciclofosfamide. Gli immunosoppressori per via orale più recenti come il fingolimod o la cladribina sono particolarmente efficaci nel ridurre la frequenza di ricadute e l’attività della malattia valutata con la RM, così come la progressione della disabilità, ma sarà necessario verificarne nella pratica clinica l’efficacia e i possibili effetti collaterali. La terapia sintomatica può migliorare alcuni disturbi come spasticità, tremore, atassia cerebellare, fatica, problemi sfinterici. La fisiochinesiterapia ha particolare rilevanza, ma deve sempre essere stabilito un programma individuale con specifici obiettivi.