
lisosoma
Ciascuno degli organuli presenti nel citoplasma con funzione digestiva dei componenti macromolecolari cellulari da eliminare. I l. appaiono come corpi rotondeggianti che si sviluppano dall’estremità distale dell’apparato del Golgi, delimitati alla periferia da una membrana simile a quella del reticolo endoplasmatico, del quale rappresentano una differenziazione. Contengono una grande varietà di enzimi idrolitici, detti idrolasi acide perché la loro azione si svolge in maniera ottimale a un pH di circa 5, il pH interno dei lisosomi. Le idrolasi sono sintetizzate nel reticolo endoplasmatico e rielaborate all’interno dell’apparato del Golgi, dal quale sono trasportate ai l. mediante speciali vescicole rivestite. I l. hanno una morfologia eterogenea che riflette un’ampia serie di funzioni digestive differenti, quali la distruzione programmata delle cellule durante l’embriogenesi, la distruzione di microrganismi fagocitati, la nutrizione cellulare. Per questo è bene considerare i l. come una serie di organuli distinti che hanno come caratteristica comune il fatto di avere un elevato contenuto di enzimi idrolitici.
Si distinguono due classi di l.: i l. primari, quelli neoformati, che non hanno perciò un substrato da digerire, e i l. secondari, sacchi membranosi con morfologia diversa e che contengono substrati da digerire ed enzimi idrolitici. I l. secondari derivano da fusioni di l. primari con diversi substrati racchiusi entro membrane, per cui la morfologia dei l. secondari varia in tanti modi quanti ne sono necessari per assumere e concentrare substrati diversi. A causa della diversa morfologia a questi sono dati nomi particolari: a) vacuoli digestivi, che derivano dalla fagocitosi di grosse particelle come i batteri; b) corpi multivescicolari, sacchi membranosi che contengono vescicole di circa 50 nm di diametro; c) vacuoli autofagici, l. che contengono e digeriscono membrane o organuli cellulari come i mitocondri.
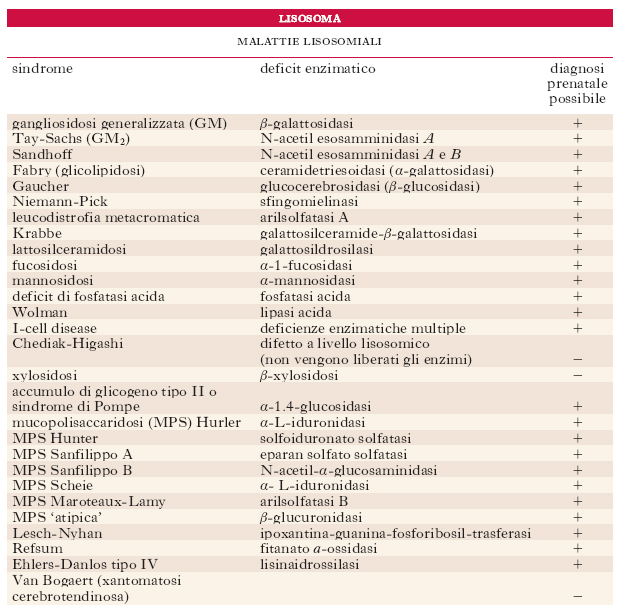
Le malattie lisosomiali rappresentano entità morbose diverse (per es., le tesaurismosi), di natura spesso ereditaria, che presentano, come carattere comune, una congenita mancanza o carenza di uno o più enzimi responsabili della degradazione dei glicolipidi o dei fosfolipidi, normalmente presenti nei lisosomi. In tabella se ne dà l’elenco completo, con le relative carenze enzimatiche che le producono e la possibilità o meno, per ognuna di esse, di una diagnosi prenatale.