
fibrosi cistica
fibrosi cistica
Malattia genetica a carattere autosomico recessivo che si manifesta clinicamente nel quadro dell’omozigosi. È la forma di patologia ereditaria più frequente con prognosi infausta.
Base eziopatogenetica del quadro morboso
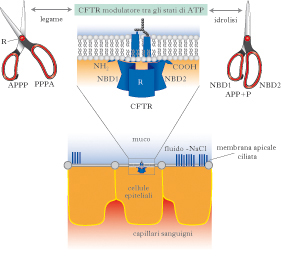
Il gene interessato codifica una proteina chiamata CFTR (Cystic Fibrosis Transmembrane conductance Regulator) la cui funzione consiste nel trasportare cloro e sodio attraverso le diverse membrane cellulari (membrana apicale delle cellule epiteliali delle vie aeree, dell’intestino, del pancreas, delle ghiandole sudoripare, dei vasi deferenti). Il gene responsabile è localizzato sul braccio lungo del cromosoma 7. Il danno biochimico si genera per un’alterata secrezione delle cellule epiteliali e riguarda in partic. gli stessi ioni cloro: ne deriva un aumentato riassorbimento di acqua e sodio. Le conseguenze si caratterizzano per un trasporto alterato del cloro a livello della membrana di cellule che formano le ghiandole a secrezione esterna. Viene secreto un muco denso, vischioso, che tende a persistere in sede. Negli organi interessati, le secrezioni determinano ostruzione dei dotti principali, provocando l’insorgenza di gran parte delle manifestazioni cliniche tipiche della malattia, come la comparsa di infezioni polmonari ricorrenti, insufficienza pancreatica, steatorrea e relativa malnutrizione, epatopatia cronica fino a cirrosi, occlusioni intestinali, alterata fertilità nel maschio.
Aspetti clinici e di diagnosi
Gli aspetti clinici sono spesso estesi a vari organi e apparati, ma in genere il quadro è dominato da un progressivo coinvolgimento del polmone. Già durante i primi mesi di vita compare tosse persistente, secca, che ricorda la pertosse. Sono sostanzialmente sempre presenti broncospasmi e aumento della frequenza respiratoria. I batteri si installano in questo particolare microambiente respiratorio e l’infezione bronchiale, cronica, recidivante, determina una progressiva distruzione del parenchima polmonare. Una grave conseguenza è l’instaurarsi di un danno anatomico irreversibile (bronchiectasia). La natura dell’espettorato assume ben presto i caratteri della secrezione purulenta e il danno istologico viene probabilmente a manifestarsi sia per la presenza di agenti patogeni sia per la stessa reazione del sistema immunitario. Il danno del pancreas, non costante, ma presente in circa i 4/5 dei soggetti con f. c. è definito da un ristagno delle secrezioni pancreatiche: nei dotti si formano cisti e si genera una fibrosi periduttale. Il risultato è un mancato assorbimento dei grassi (dal quale deriva la steatorrea) e non assorbimento delle vitamine liposolubili A, D, E e K. Il progredire nel tempo del danno pancreatico induce anche un diabete mellito, per insufficiente produzione di insulina. Per arrivare alla diagnosi si esegue il test del sudore nel neonato: se i valori di cloro sono maggiori di 60 meq/L nel sudore allora il test risulta positivo. Altra indagine di screening si può eseguire sul meconio. In relazione alla trasmissibilità del gene per fare diagnosi prenatale, nelle famiglie a rischio, si attua il prelievo dei villi coriali.
Implicazioni assistenziali e prognosi
La f. c. è un importante problema assistenziale e la prognosi non consente, in genere, un esito favorevole oltre i 50 anni. Usando adeguati sistemi di alimentazione, enzimi pancreatici sostitutivi, fisioterapia respiratoria, trattamenti antibiotici ciclici a dosi di farmaco adeguate, la qualità della vita è migliorata ma risulta comunque bassa e implica notevole impegno assistenziale, anche sotto il profilo psicologico. Per aiutare i malati sono stabilite norme di legge che consentono la fornitura, in Italia, di materiale medico gratuito, con i vari approcci curativi necessari e la riabilitazione domiciliare.
I meccanismi molecolari della fibrosi cistica
La fibrosi cistica è una delle malattie genetiche letali più comuni nella popolazione caucasica; viene trasmessa con modalità autosomica recessiva ed è causata da mutazioni nel gene CFTR, dalle quali dipende la produzione della corrispettiva forma alterata della proteina canale transmembrana del cloro. I pazienti affetti da fibrosi cistica in Europa e nell’America settentrionale sono più di 70.000; in Italia viene diagnosticata in un neonato ogni 2.700, e si stima che i portatori sani siano circa 2 milioni e mezzo. La fibrosi cistica si presenta come una malattia sistemica che ha effetti sulle ghiandole esocrine di polmoni, fegato, pancreas, e intestino, causando malattie croniche dell’apparato respiratorio, insufficienza pancreatica esocrina, disturbi dell’apparato gastro-intestinale. Sebbene l’aspettativa di vita dei pazienti affetti da fibrosi cistica sia aumentata negli ultimi decenni, la morte, causata da insufficienza respiratoria, sopraggiunge mediamente intorno ai 37 anni di età con sopravvivenza massima poco sopra i 50 anni.
Meccanismo
Il locus della mutazione nel gene CFTR (Cystic Fibrosis Transmembrane Conductance Regulator gene) è stato localizzato nel 1989 sul braccio lungo del cromosoma 7; il gene è costituito da 250 kb di DNA genomico, contiene 27 esoni e produce un mRNA di 6,5 kb. Sequenziando il DNA di persone affette sono state individuati più di 750 alleli patogeni e più di mille diverse mutazioni. Il gene CFTR codifica una proteina costituita da 1.480 amminoacidi, localizzata nella membrana apicale delle cellule degli epiteli. La proteina CFTR è un canale transmembrana del cloro, attivato dalla proteina chinasi A mediante AMP ciclico, con la funzione di regolare gli scambi elettrolitici tra l’interno e l’esterno delle cellule di molte ghiandole dell’organismo. La sua alterazione causa anomalie del trasporto di ioni cloro attraverso le membrane cellulari e, in generale, della produzione delle secrezioni esocrine dell’organismo, che risultano dense e viscose, causando un danno progressivo degli organi coinvolti. Si ritiene che il più grave effetto dovuto all’accumulo di muco sia la creazione di un microambiente favorevole alle infezioni polmonari croniche da parte di batteri come Staphylococcus aureus e Pseudomonas aeruginosa, che sono la causa di morte più frequente per i pazienti affetti da fibrosi cistica.
Tipi di mutazioni
Nella popolazione caucasica la mutazione più frequente del gene CTFR consiste nella delezione dell’amminoacido fenilalanina, presente nella posizione 508 della proteina normale. Questa mutazione è chiamata ΔF508 ed è responsabile della maggior parte dei casi di fibrosi cistica; a seconda della regione geografica, circa il 50÷70% degli individui affetti sono omozigoti. La mutazione (ΔF508 è molto antica e probabilmente si è diffusa nelle popolazioni europee grazie alla selezione a favore dell’eterozigote; poiché il gene della fibrosi cistica codifica un canale di membrana per il cloro, la cui presenza è necessaria affinchè la Salmonella typhi entri nelle cellule epiteliali, gli eterozigoti potrebbero essere relativamente resistenti alla febbre tifoide.
Finora sono state identificate più di un migliaio di altre mutazioni nel gene CFTR, la cui frequenza relativa è molto variabile in relazione all’area geografica. Le mutazioni nel gene CFTR sono catalogate in cinque classi, in base al meccanismo con cui alterano la funzione della proteina. Le mutazioni di classe I portano alla interruzione prematura della trascrizione, che ha come risultato un trascritto CFTR troncato e instabile, oppure un’espressione del tutto assente di CFTR. Le mutazioni missense (classe II) danno origine a un codone che specifica un amminoacido diverso da quello originario, o deleto, come nella mutazione (ΔF508. Queste mutazioni causano un avvolgimento errato della proteina che porta al trattenimento della proteina stessa nel reticolo endoplasmatico, e alla sua prematura degradazione. Le mutazioni di classe III e IV sono meno gravi e hanno come risultato la ridotta capacità di CFTR di secernere o di condurre gli ioni cloro, mentre le mutazioni di classe V causano uno splicing anomalo, che riduce la quantità di proteina funzionale prodotta.
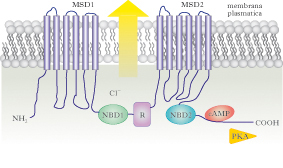
La proteina CFTR
CFTR è un membro della superfamiglia di proteine del trasporto ABC (ATP-Binding Cassette). Queste proteine sono regolate dall’AMP ciclico, e hanno in comune la funzione di trasporto di soluti, dagli ioni organici ai carboidrati, attraverso la membrana cellulare. Le mutazioni nei geni ABC sono associate a molte malattie genetiche, tra cui la sindrome di Dubin-Johnson e la degenerazione della macula. Strutturalmente la proteina CFTR è un canale ionico formato da quattro domini disposti in due metà simmetriche, ciascuna comprendente un dominio transmembrana (MSD1 e MSD2) composto di sei segmenti, e un dominio NBD (Nucleotide Binding Domain) citoplasmatico, con proprietà di idrolisi dell’ATP. Le due metà della proteina sono collegate da un dominio regolativo che, in accordo con i domini NBD, modula l’apertura del canale del cloro. Quando l’ATP è legato ai due domini NBD, il canale è aperto. L’idrolisi di una delle due molecole di ATP distrugge l’interazione NBD1-NBD2 e chiude il canale del cloro, interrompendo il flusso degli ioni. La proteina CFTR, dopo la sintesi, viene sottoposta a un processo di maturazione post-traduzionale che avviene nel reticolo endoplasmatico e richiede la partecipazione di un complesso di proteine chaperon, tra cui Hsp90, che controllano il corretto avvolgimento della proteina ed, eventualmente, come nel caso della mutazione ΔF508, la indirizzano verso un pathway degradativo.