epilessia
epilessia
Sindrome caratterizzata da manifestazioni motorie, sensoriali, psichiche e neurovegetative polimorfe, in forma di crisi che si ripetono nel tempo, dipendenti da scariche parossistiche neuronali, provenienti da differenti regioni cerebrali.
Eziopatogenesi
Seppure per la maggior parte di e. non è individuabile una causa, quando questa è nota di maggior fraquenza sono i traumi, le infezioni cerebrali e meningee e i loro esiti, l’alcolismo, le neoplasie, alcune anomalie vascolari. Si ritiene che, indipendentemente dalle cause, gran parte delle crisi abbia origine da un insieme di neuroni (focolaio) con bassa soglia di eccitabilità; lo stimolo iniziale si propaga attraverso i circuiti neuronali circostanti.
Classificazione
Le e. generalizzate idiopatiche includono diverse sindromi, nelle quali svolgono spesso un ruolo fattori scatenanti, quali l’alterazione del ritmo sonno-veglia, eventi stressanti, stimolazione luminosa intermittente, ecc. Comprendono: le convulsioni neonatali benigne (frequenti crisi miocloniche o di apnea); il piccolo male a tipo assenza infantile e giovanile, che rappresenta una delle forme più frequenti, con improvvisa sospensione della coscienza di breve durata (5÷10 s); l’e. mioclonica giovanile; l’e. con crisi di grande male al risveglio, con improvvisa perdita di coscienza, seguita da caduta a terra, con una fase tonica (arresto del respiro, spesso cianosi, contrattura dei muscoli masticatori e blocco delle mascelle, morsicatura della lingua), seguita da fase clonica, caratterizzata da scosse ritmiche agli arti. Le e. generalizzate criptogenetiche e sintomatiche sono forme gravi, con crisi molto frequenti, resistenti alla terapia. Si ricordano la sindrome di West e quella di Lennox-Gastaut. Le e. parziali comprendono quelle forme in cui la scarica che le sottende interessa una popolazione di cellule cerebrali circoscritta, dando origine a una crisi parziale semplice o complessa, o anche generalizzata se, pur avendo un inizio localizzato, sfugge poi al controllo dei meccanismi inibitori e si diffonde a entrambi gli emisferi cerebrali; i sintomi possono essere diversi, in quanto sono legati all’area della corteccia cerebrale che viene interessata dalla scarica, ma sono costanti nel singolo paziente. Un particolare tipo di crisi è quello che si verifica quando viene coinvolta la corteccia motoria, con crisi parziali semplici motorie, che iniziano con contrazioni muscolari ritmiche, limitate a un solo segmento corporeo oppure coinvolgenti in sequenza un intero lato del corpo (crisi jacksoniana con marcia). L’e. temporale, con focolaio nel lobo temporale, è costituita da sensazioni anomale varie, da movimenti automatici elementari, coscienza obnubilata. Lo stato di male epilettico rappresenta una grave complicazione di qualsiasi forma di e. ed è caratterizzato dal ripetersi, con ritmo subentrante, di accessi generalizzati, senza ripristino della coscienza nelle fasi intervallari.
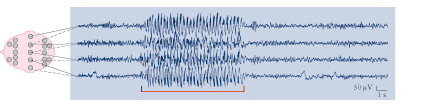
Diagnosi e terapia
La diagnosi si basa sull’osservazione dei vari sintomi e, soprattutto, sull’elettroencefalografia. L’efficacia della cura è condizionata dalla scelta dei farmaci (solitamente si associano più molecole) e dalla regolarità nella loro assunzione; i farmaci antiepilettici sono in continuo aumento: primidone, carbamazepina, valproato di sodio, gabapentin, sono stati seguiti, dalla fine del secolo scorso, da topiramato, levetiracetam e altri. Diazepam, acetazolamide, derivati della fenilsuccinamide, sono associati spesso alla terapia di base. Alla terapia chirurgica si può ricorrere nei casi di e. refrattaria al trattamento farmacologico.
L’epilessia post-traumatica
Tra le sequele immediate e tardive del trauma cranico, la più frequente è la sindrome post-concussionale. Si stima che il 25% dei pazienti con traumi chiusi con contusione cerebrale e il 50% di quelli con ferite penetranti che provocano brecce durali, sviluppino attacchi epilettici, che avvengono entro le prime 24 ore dalla lesione. Questi attacchi non si associano a prognosi negativa se confinati alle prime 24 ore, ma si accompagnano alla probabilità di sviluppare forme di e. post-traumatica se si presentano dopo il primo giorno o entro le prime due settimane. L’e. post-traumatica si distingue in precoce e tardiva. Circa il 5% dei fenomeni convulsivi si sviluppa nella settimana successiva all’evento e poi nuovamente in un 25% dei pazienti. In non più del 10% dei casi le convulsioni si manifestano dopo la prima settimana, variando molto in relazione al tipo specifico del trauma e alla severità della lesione (circa il 50% nei casi di ematoma intracerebrale e subdurale, e fino al 50% in pazienti con fratture depresse). La maggior parte degli attacchi ricorrenti compare infine entro due anni dal trauma cranico, e rappresenta la forma di e. post-traumatica tardiva. L’incidenza della e. post-traumatica tardiva aumenta in presenza di lacerazioni durali, amnesia posttraumatica superiore alle 24 ore e coma. Nel complesso, circa il 50% dei pazienti con accessi post-traumatici guarisce spontaneamente, il 25% va controllato con terapia farmacologica e l’altro 25% è resistente alle terapie. Generalmente, nella terapia di pazienti con trauma cranico si utilizzano farmaci anticonvulsivi soltanto se l'e. si rivela persistente.