
Terapia genica
Terapia genica
La terapia genica si è sviluppata, a partire dalla fine degli anni Ottanta del XX sec., grazie ai progressi compiuti nella comprensione delle basi molecolari di molte malattie umane e nella messa a punto di tecniche efficienti per il trasferimento dei geni. Questa disciplina è nata per curare alcune malattie monogeniche con ereditarietà recessiva, mediante l'inserzione, nei tessuti appropriati, di una copia normale del gene mutato. Oltre ai geni codificanti, sfrutta oggi anche il trasferimento di frammenti di DNA e di RNA con funzione enzimatica e regolatrice, e si rivolge anche al trattamento dei tumori, delle malattie cardiovascolari e dell'AIDS. Nella maggior parte dei casi, la terapia genica fa ricorso a vettori virali per il trasferimento degli acidi nucleici con funzione terapeutica; i più utilizzati sono quelli basati sui retrovirus murini, sull'adenovirus e sul virus adenoassociato (AAV). I risultati delle sperimentazioni cliniche finora compiute indicano che il trasferimento genico nell'uomo è realizzabile, ma che sono ancora molti i traguardi conoscitivi e tecnologici che devono essere raggiunti prima che la terapia genica possa contribuire in maniera significativa alla cura di molte malattie oggi intrattabili.
La rivoluzione tecnologica e scientifica prodotta dall'avvento dell'ingegneria genetica, cioè dall'acquisizione della capacità di modificare in modo mirato le proprietà ereditabili degli organismi viventi, ha indotto molti gruppi di ricerca, a partire dalla fine degli anni Ottanta, ad applicare queste nuove tecnologie alla cura di diverse patologie. Com'era in un certo senso naturale, il primo obiettivo considerato è stata la cura delle malattie ereditarie monogeniche recessive: se si fossero potute correggere, per esempio, le specifiche mutazioni patologiche nel gene della catena β dell'emoglobina, sarebbe stato possibile curare una malattia ereditaria grave e diffusa quale la β-talassemia. Più modestamente, dal momento che questa patologia ha un'ereditarietà di tipo recessivo, cioè è sufficiente un solo allele normale del gene per conferire uno stato di normalità, il medesimo risultato terapeutico si sarebbe potuto ottenere inserendo una copia attiva ed efficacemente regolata del gene 'sano' nelle cellule portanti la mutazione. Alla luce delle difficoltà che si incontrano nell'ottenere modifiche mirate in un punto preciso del genoma delle cellule di mammifero, la terapia genica delle malattie ereditarie è nata proprio sfruttando questa seconda opzione.
Negli anni successivi ci si è resi conto che questo approccio poteva essere esteso ad altre patologie, molto più diffuse nella popolazione rispetto alle malattie ereditarie monogeniche. Infatti, il repertorio di possibili geni terapeutici si estende al di là di quelli con funzione sostitutiva e comprende geni o frammenti di DNA o RNA con funzioni di regolazione dell'attività cellulare o di modulazione del sistema immunitario. Di conseguenza, lo spettro di potenzialità terapeutiche della terapia genica si rivolge a molte malattie acquisite, quali le neoplasie, le malattie cardiovascolari e l'infezione da HIV.
Parallelamente all'allargamento delle possibilità terapeutiche della terapia genica, negli ultimi anni si è assistito a una presa di coscienza dell'esistenza di serie difficoltà tecniche e concettuali che rendono ancora più problematica l'applicazione clinica su vasta scala di questa tecnologia. Queste difficoltà sono apparse evidenti analizzando i risultati preliminari ottenuti dallo studio delle ormai diverse migliaia di pazienti trattati mediante trasferimento genico. Verranno qui delineate le principali strategie utilizzate per la terapia genica, analizzando i possibili geni terapeutici, le tecniche per il loro il trasferimento, i primi risultati conseguiti dalla sperimentazione clinica e i problemi tecnici, etici e sociali ancora da superare.
Tecniche per il trasferimento genico
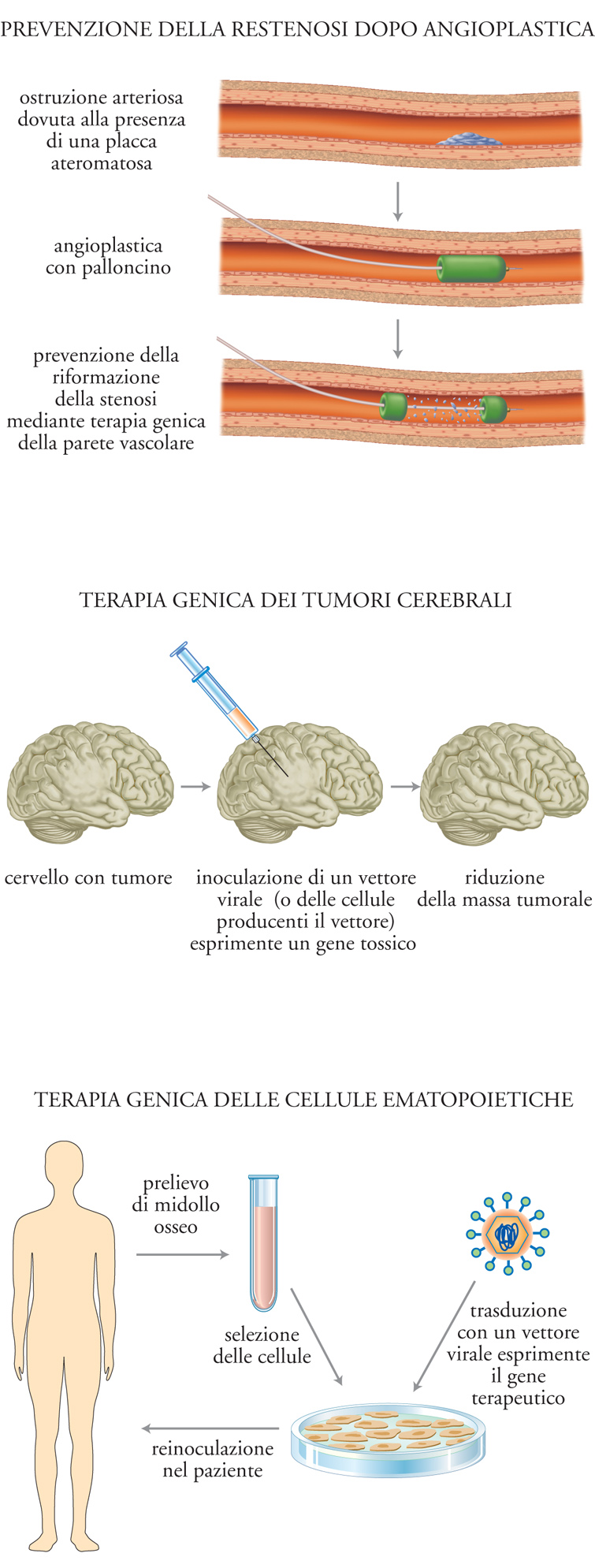

Il successo della terapia genica è strettamente dipendente dall'efficienza con cui il materiale genetico estraneo viene introdotto nel genoma delle cellule sottoposte al trattamento. Esistono due tipi di applicazioni: il primo si propone di trattare le cellule bersaglio dopo averle prelevate dal paziente e coltivate in vitro; dopo il trattamento, le cellule ingegnerizzate vengono reinfuse nel paziente. Tale trattamento ex vivo è appropriato, per esempio, per il trasferimento genico nei precursori midollari o negli epatociti. Le applicazioni del secondo tipo prevedono invece trattamenti realmente in vivo, per esempio per il trasferimento genico nel muscolo, nel cervello o nelle cellule della parete arteriosa. Una rappresentazione schematica di queste diverse possibilità di terapia genica è indicata nella fig. 2. Gli esempi sono tratti da alcune sperimentazioni cliniche che verranno descritte e commentate in seguito. Le principali metodiche disponibili per il trasferimento genico, i loro vantaggi e gli svantaggi sono presentati nella tab. 1.
Aggiunta di DNA nudo
La semplice aggiunta di DNA nudo, sotto forma di plasmide, al milieu extracellulare è un sistema di regola poco efficiente per ottenere l'ingresso del DNA all'interno delle cellule. Fanno eccezione alcuni tessuti, quali il muscoloscheletrico e il cardiaco, le cui cellule sono in grado di endocitare attivamente il DNA, che giunge al nucleo e viene espresso. Relativamente più efficiente è invece la somministrazione di corti frammenti di DNA (oligonucleotidi formati da 15-30 nucleotidi), che sono in grado di entrare nella cellula se somministrati ad alta concentrazione. La sequenza di queste molecole può essere disegnata in maniera tale da risultare complementare a quella degli mRNA dei geni cellulari o virali. Queste molecole antisenso, appaiandosi con il loro bersaglio, ne stimolano la degradazione a opera di nucleasi cellulari che riconoscono gli ibridi DNA/RNA. Questo tipo di approccio è stato sfruttato largamente in laboratorio per inibire sperimentalmente la funzione di diversi geni. Gli oligonucleotidi più efficaci portavano modifiche chimiche a livello dello scheletro dei legami fosfodiesterici del DNA, che li rendevano resistenti alla degradazione e ne miglioravano le proprietà farmacocinetiche. Più recentemente, tuttavia, l'utilizzo di oligonucleotidi è stato largamente sostituito dagli approcci basati sulle tecnologie dell'interferenza genica basata sull'RNA.
Metodi chimico-fisici
Nonostante il fatto che i sistemi di trasferimento genico più utilizzati siano quelli basati sui vettori virali, almeno due motivi suggeriscono che i sistemi di trasferimento non virali risulteranno in futuro quelli preferiti: la sicurezza e la facilità di produzione. Un sistema di trasferimento genico totalmente sintetico elimina il rischio di contaminazione da parte di virus in grado di replicarsi in modo autonomo, oltre a quello legato agli effetti tossici causati dall'infezione virale stessa. Inoltre, la produzione di un composto sintetico risulterà certamente più semplice dell'utilizzo dei sistemi biologici disponibili per la produzione dei vettori virali. Tuttavia, attualmente, questi vantaggi del trasferimento genico non virale sono controbilanciati in larga misura dalla loro relativa inefficienza.
Tra i metodi chimici di trasferimento genico, il più studiato è la lipofezione, che utilizza i lipidi cationici e i liposomi. I lipidi cationici instaurano interazioni ioniche con il DNA formando complessi; i liposomi formano delle vere e proprie vescicolette con struttura lamellare che circondano il DNA. In entrambi i casi, il DNA viene trasportato in maniera efficiente attraverso la membrana citoplasmatica e internalizzato negli endosomi della cellula. Le particelle formate dall'interazione dei lipidi con il DNA possono essere veicolate su determinati recettori cellulari mediante l'introduzione di specifici ligandi peptidici. Esempi di tali ligandi sono l'emoagglutinina del virus Sendai, in grado di aumentare l'efficienza della fusione del liposoma con la membrana cellulare, o i peptidi che portano la sequenza amminoacidica Arg-Gly-Asp (RGD), in grado di legarsi in maniera specifica ai recettori integrinici della cellula. Queste metodiche sono ancora largamente inefficaci in termini quantitativi, in quanto il lipide trasportatore riversa all'interno della cellula molti milioni di molecole di DNA che si vengono a trovare all'interno delle vescicole endolisosomiali, dove per la maggior parte sono degradate; solo una piccola frazione di esse sfugge alla degradazione, viene portata nel nucleo ed è espressa. Una porzione ancora minore viene integrata in maniera casuale nel genoma.
Il trasferimento del DNA può avvenire anche formando complessi DNA/proteine: tali complessi sfruttano le proprietà di alcune proteine di interagire con recettori di membrana specifici di determinate cellule. Il complesso viene poi internalizzato nella cellula tramite un meccanismo di endocitosi mediata dal recettore. Per esempio, l'asialoglicoproteina si lega in maniera specifica a un recettore sugli epatociti; la transferrina utilizza recettori presenti in diversi tipi cellulari. Analogamente ai metodi di lipofezione, anche con questa metodica i livelli di espressione del DNA trasferito risultano modesti e transitori. Infatti, anche in questo caso il complesso DNA/proteina rimane per la maggior parte intrappolato negli endosomi della cellula e viene perciò degradato rapidamente. Tuttavia, il trattamento contemporaneo con agenti endosomolitici (come l'aggiunta di virus inattivati, quali l'adenovirus, che fisiologicamente sono in grado di uscire dagli endosomi) permette di aumentare l'efficacia di questa strategia di diverse migliaia di volte.
Metodi fisici
Tra i metodi fisici, appare promettente la metodica del bombardamento delle cellule con microscopiche particelle ricoperte di DNA. La variante più usata impiega vere e proprie pistole che sparano ad altissima velocità nei tessuti particelle di oro rivestite del gene che interessa (gene gun). Queste particelle sono in grado di attraversare la membrana citoplasmatica e quella nucleare rilasciando il DNA nel nucleo delle cellule del tessuto bersaglio. Questa metodologia, che deriva da analoghe tecniche utilizzate da anni per il trasferimento genico nelle piante, trova applicazione per la terapia genica in vivo di tessuti accessibili quali quello cutaneo, particolarmente con lo scopo di trasferire geni che codificano proteine in grado di stimolare il sistema immunitario. A livello del derma, il DNA viene assunto ed espresso da cellule con funzione di APC (Antigen presenting cell) e in grado, quindi, di fornire una specifica e forte stimolazione sia dell'immunità cellulare (mediata dai linfociti T) sia di quella anticorpale (mediata anche dai linfociti B). Ne consegue che l'applicazione di questa tecnologia appare indicata per ottenere la vaccinazione contro agenti patogeni, di cui si utilizzano singoli geni in grado di stimolare una risposta protettiva (vaccinazione a DNA).
Vettori virali
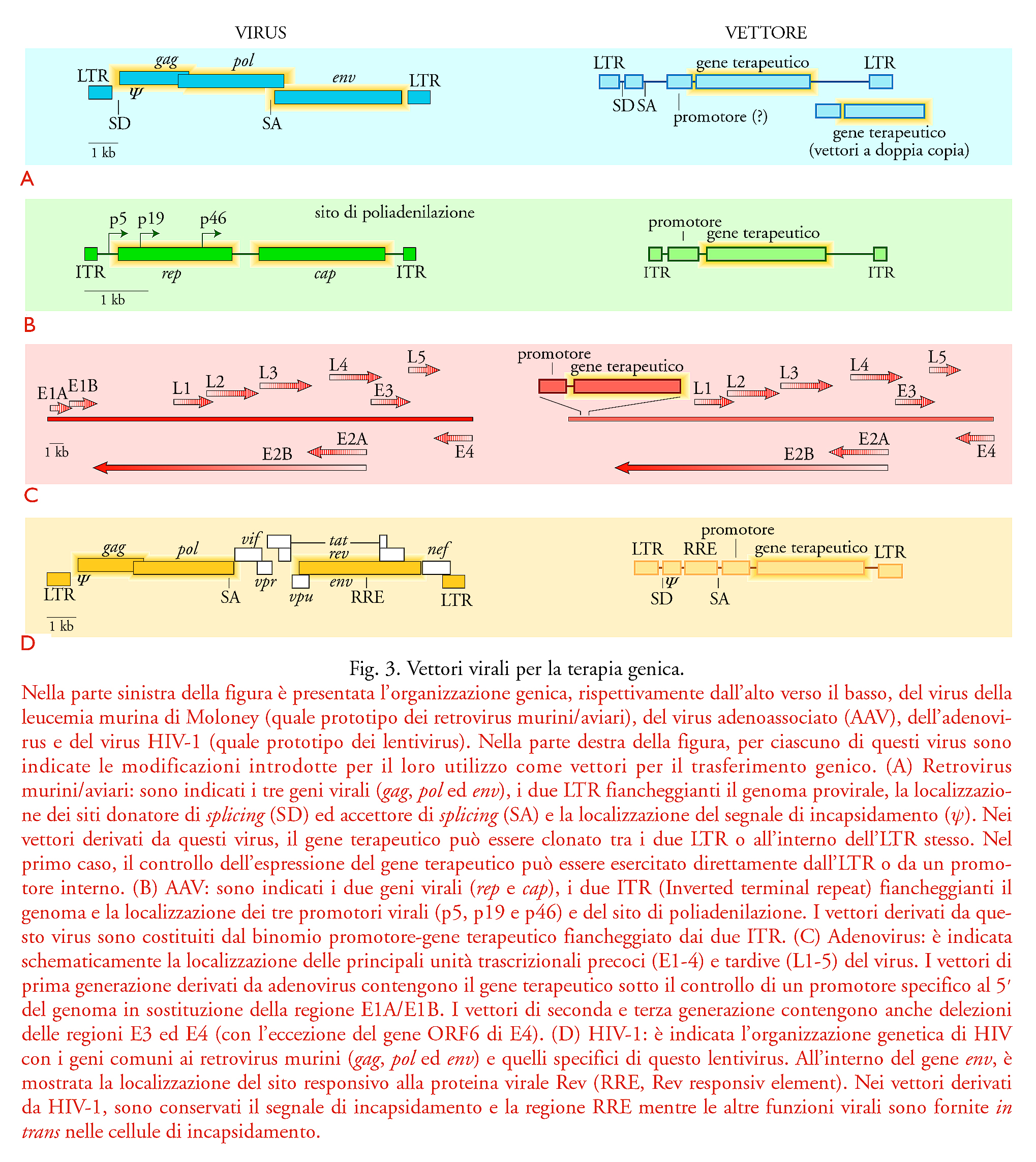
Le metodologie di trasferimento genico maggiormente usate per la terapia genica sono basate sull'utilizzo di vettori virali e sfruttano le proprietà fisiologiche dei virus di infettare le cellule bersaglio con alta efficienza, interagendo con recettori cellulari specifici. Nella fig. 3 è presentata in maniera schematica l'organizzazione genica dei quattro principali tipi di virus utilizzati come vettori: retrovirus animali, virus adenoassociati, adenovirus e lentivirus. I principali vantaggi e svantaggi di questi vettori per il trasferimento genico sono anche riassunti nella tab. 1.
I vettori virali di gran lunga più impiegati si basano sui retrovirus aviari e murini. Questi vettori sono privilegiati per la loro relativa semplicità genetica, perché possono infettare con alta efficienza una vasta gamma di tipi cellulari, e perché il loro ciclo biologico contempla una forma di DNA provirale che si integra in maniera stabile nel genoma della cellula ospite. Il genoma della forma provirale di un tipico retrovirus è costituito da due lunghe sequenze terminali ripetute (LTR, Long terminal repeats), che si vengono a formare durante il processo di trascrizione inversa, e da tre geni fondamentali, che codificano, rispettivamente, le proteine del core nucleoproteico (gene gag), le proteine del pericapside (gene env) e la trascrittasi inversa/integrasi/proteasi (gene pol) (fig. 3A). Il provirus integrato si comporta come un qualsiasi gene cellulare: la sua replicazione coincide con il processo di trascrizione, che è compiuto dall'RNA-polimerasi II cellulare e regolato dai fattori di trascrizione cellulari che si legano all'LTR all'estremità 5′ del provirus. Il trascritto inizia a livello dell'LTR al 5′ e termina a livello dell'LTR al 3′ del provirus; esso rappresenta sia il genoma virale che viene incluso nella particella virale, sia l'mRNA che viene utilizzato come stampo per la traduzione delle proteine virali. Queste ultime vengono tradotte direttamente dall'mRNA genomico (nel caso di Gag e Pol) o dopo processamento di questo RNA (nel caso di Env).
Un vettore retrovirale viene ricavato da questo scheletro lasciando inalterati i due LTR e un'unica regione all'estremità 5′ del gene gag che contiene il segnale per l'inclusione nella particella virale (regione ψ), mentre tutto il resto del genoma può essere sostituito dal gene terapeutico o da altri geni di interesse. Sono state proposte diverse varianti per la costruzione di vettori retrovirali. Queste includono la scelta dell'LTR del retrovirus (diversi tipi di retrovirus murini e aviari hanno tropismi cellulari diversi e regolano l'espressione del genoma con maggiore o minore efficienza); la possibile introduzione di un promotore appropriato, per esempio specifico per un certo tipo di tessuto, a monte del gene terapeutico; la mutazione o la conservazione dei siti di splicing del retrovirus; l'introduzione di un secondo gene che permetta la selezione delle cellule trasdotte, a sua volta controllato da un promotore interno.
Un'opzione alternativa alle precedenti è la clonazione del gene terapeutico nell'LTR al 3′ del vettore, in modo che il processo di trascrizione inversa lo copi anche a livello dell'LTR al 5′, così da duplicare il suo numero di copie. Lo sviluppo di tutte queste varianti mira a ottenere sia vettori sempre più efficienti nella capacità di trasdurre geni terapeutici, sia maggiori livelli di espressione del gene terapeutico nelle cellule trasdotte.
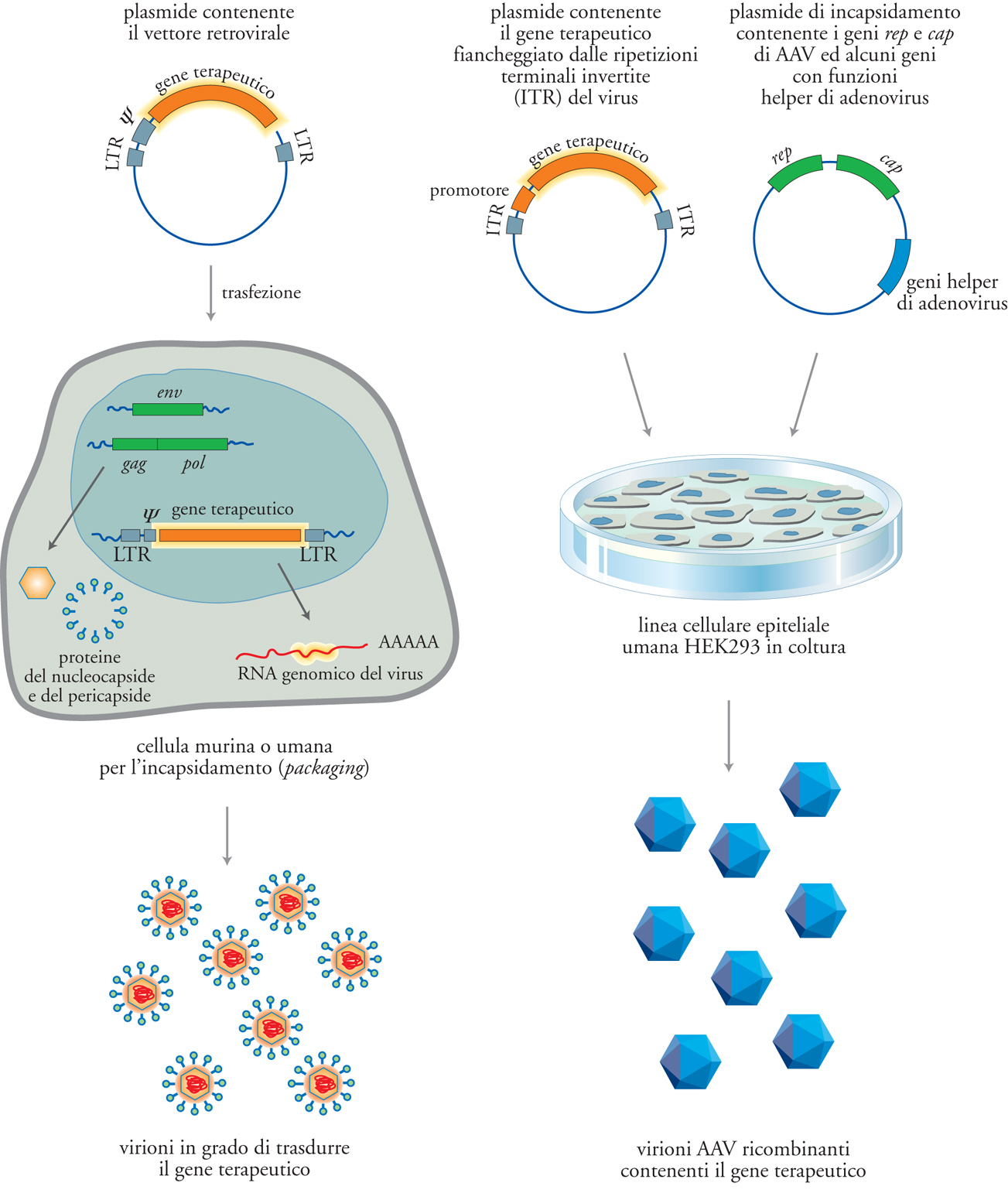
Una volta costruito il vettore retrovirale di scelta sotto forma di DNA plasmidico, esso viene trasferito in laboratorio in una linea cellulare, solitamente murina, utilizzata per la produzione dei virioni ricombinanti (packaging cell line). Le cellule di queste linee contengono, in diverse localizzazioni del genoma, i geni retrovirali gag, pol ed env e producono le relative proteine (fig. 4). Il DNA plasmidico trasferito trascriverà il genoma del vettore, il quale, contenendo la regione ψ, riconoscerà in maniera specifica queste proteine e sarà incluso nel virione. Ne risulterà la produzione di una particella retrovirale completa che porterà, quale genoma, quello del vettore. Questa particella è in grado di infettare con alta efficienza il tessuto bersaglio e di trasferire l'RNA genomico del vettore, che viene retrotrascritto in maniera corretta, e si integra in forma provirale nei cromosomi delle cellule ospiti. Il vettore, a questo punto, non risulta più in grado di replicarsi, ma funziona come un gene aggiunto al patrimonio genetico dell'ospite. La maggioranza delle sperimentazioni cliniche attualmente in corso fa uso di vettori retrovirali murini o aviari, sebbene alcuni svantaggi ne rendano problematico l'utilizzo per alcune applicazioni. Uno di questi svantaggi è il basso titolo con cui i vettori retrovirali possono essere prodotti: al massimo 1∙107 ÷ 1∙109 particelle virali infettive/ml, a seconda della specificità del recettore con cui il vettore interagisce.
Un altro problema è rappresentato dal fatto che l'integrazione della forma provirale all'interno del genoma della cellula ospite necessita di almeno un ciclo di replicazione cellulare, in quanto il complesso di preintegrazione non è in grado di essere trasportato attraverso la membrana nucleare intatta. Questo limita l'utilizzo di tali vettori nelle cellule che non si replicano attivamente, quali i neuroni del sistema nervoso centrale o i precursori staminali ematopoietici, in cui la stimolazione proliferativa ‒ almeno in vitro e nelle condizioni sperimentali attualmente utilizzate ‒ coincide, quasi inevitabilmente, con uno stimolo differenziativo e con la conseguente perdita delle proprietà staminali stesse. Questa difficoltà potrebbe essere superata con l'utilizzo di vettori retrovirali basati sulla famiglia dei lentivirus, usando come prototipo HIV-1: infatti il genoma di questi virus è in grado di essere trasportato nel nucleo e di integrarsi nel DNA dell'ospite in assenza di replicazione delle cellule infettate, grazie a due proteine virali, Vpr e MA, che hanno la proprietà di trasportare il complesso di preintegrazione, formatosi nel citoplasma, attraverso la membrana nucleare. Questi vettori sono costruiti in maniera concettualmente analoga a quella dei retrovirus murini e aviari, in quanto contengono il gene terapeutico, sotto il controllo di un promotore appropriato, e il sito per l'inclusione nel nucleocapside, fiancheggiati dai due LTR virali (fig. 3D). I vettori contengono anche la regione RRE di HIV-1, indispensabile per il legame alla proteina Rev e il conseguente trasferimento dell'RNA dal nucleo al citoplasma nelle cellule utilizzate per la produzione dei virioni ricombinanti. Tutte le funzioni virali per la regolazione e la produzione delle particelle virali e accessorie sono fornite in trans nelle cellule utilizzate per la produzione dei virioni ricombinanti grazie all'espressione dei geni di HIV-1 da parte di due plasmidi che sono trasfettati simultaneamente con il vettore. Ovviamente, l'utilizzo di vettori basati su HIV-1 solleva l'importante preoccupazione della loro sicurezza. Un ulteriore grave problema che limita l'utilizzo dei vettori retrovirali e lentivirali è legato alla possibilità che il processo di integrazione nel genoma della cellula trasdotta, che avviene casualmente, possa portare all'inattivazione di un gene oncosoppressore o all'attivazione di un oncogene e possa quindi causare la trasformazione in senso neoplastico della cellula, un evento in linea teorica estremamente improbabile ma purtroppo recentemente osservato nel corso di una sperimentazione clinica.
Nel caso in cui l'integrazione nel genoma della cellula trasdotta non sia opportuna, o comunque non sia richiesta ‒ come nella terapia genica dei tumori, nelle vaccinazioni, nella terapia della parete vascolare per la prevenzione della restenosi ‒ una classe molto interessante di vettori è rappresentata dagli adenovirus. I vettori adenovirali hanno molte caratteristiche interessanti che li rendono appropriati per il trasferimento genico in vivo. Essi possono contenere ampie regioni di DNA terapeutico (fino a circa 30 kb nei vettori dell'ultima generazione), sono in grado di infettare molti tipi cellulari ‒ incluse le cellule quiescenti ‒ con grande efficienza (spesso vicina al 100% negli esperimenti di laboratorio) e possono essere prodotti con titoli molto elevati. Sono attualmente disponibili diverse generazioni di vettori adenovirali, che si differenziano per le diverse porzioni di genoma virale delete per fare spazio al gene terapeutico e per rimuovere funzioni potenzialmente dannose (fig. 3C). I vettori di prima generazione contenevano il gene terapeutico nella porzione al 5′ del genoma, dove erano delete le regioni che codificano le proteine E1A e E1B. Questi geni erano forniti in trans dalle cellule utilizzate per la formazione delle particelle virali. Vettori più recenti, sviluppati con lo scopo di ridurre il livello di infiammazione e di riconoscimento immunologico causato dall'adenovirus, contengono anche delezioni nelle regioni E3 ed E4. Infine, i vettori adenovirali più recenti comprendono virus in cui la maggior parte del genoma è deleta, e le proteine per la loro replicazione sono fornite in trans nelle stesse cellule in cui le particelle virali sono prodotte. A questo proposito va osservato che la delezione di tutti i geni virali non necessariamente migliora l'efficacia, dal momento che alcuni di questi geni possono risultare vantaggiosi per il vettore. Per esempio, la regione E3 codifica una proteina di 19 kDa che protegge il virus, e presumibilmente il vettore, dal riconoscimento da parte del sistema immunitario.
Una ulteriore classe di vettori virali è costituita da quelli basati sui virus adenoassociati (AAV). Questi sono piccoli virus a DNA a singolo filamento, che richiedono funzioni ausiliarie cellulari per la loro replicazione, e che sono caratterizzati dalla proprietà di integrarsi con alta frequenza nel genoma delle cellula ospite, anche in assenza di proliferazione cellulare. Inoltre, il genoma virale del virus selvatico si integra in maniera preferenziale in una regione del cromosoma 19 (banda 19q13.3) in virtù di meccanismi molecolari non ancora chiariti. Questa proprietà, qualora potesse essere mantenuta anche nei vettori ricombinanti, potrebbe costituire un grosso vantaggio per la terapia genica, in quanto eviterebbe il rischio della mutagenesi inserzionale, che caratterizza sia i retrovirus sia tutti gli altri sistemi di trasferimento genico che prevedono l'inserzione inevitabilmente casuale del DNA esogeno nel genoma della cellula ospite.
Il genoma virale (fig. 3B) degli AAV è costituto da due sequenze ripetute in orientamento invertito alle estremità (ITR, Inverted terminal repeat) e da due geni (rep e cap), che codificano, rispettivamente, proteine coinvolte nella replicazione (Rep) e nell'assemblaggio del capside virale (Cap). I vettori adenoassociati contengono unicamente le sequenze ITR terminali (165 nucleotidi) che fiancheggiano il gene terapeutico, mentre le funzioni di rep e di cap sono prodotte mediante trasferimento genico di un plasmide che codifica questi geni nelle cellule utilizzate per la produzione delle particelle ricombinanti. Analogamente ai vettori retrovirali, da queste cellule viene prodotto un virione infettivo che contiene il genoma ricombinante con il gene terapeutico (fig. 4 a destra). I virioni prodotti, a differenza dei retrovirus, possono anche essere purificati e concentrati con alta efficienza, in modo da ottenere preparazioni con un titolo infettivo molto elevato. Tuttavia, le caratteristiche molecolari della replicazione degli AAV, e in particolare le funzioni cellulari indispensabili per la sintesi del filamento complementare al genoma virale, l'ingresso nel nucleo e l'integrazione nel genoma, sono a tutt'oggi poco conosciute. Infine, anche gli herpesvirus umani sono potenzialmente dei vettori utili per la terapia genica, soprattutto in virtù della loro caratteristica di instaurare un'infezione latente nell'organismo. In quest'ottica, risulta particolarmente interessante il virus dell'Herpes simplex di tipo 1 (HSV-1) per la sua capacità di infettare e permanere indefinitamente nelle cellule del sistema nervoso centrale. Tuttavia, la grande dimensione del loro genoma (150÷200 kb), che ne limita l'uso in laboratorio, e la scarsa comprensione dei loro meccanismi di patogenicità, ne prevengono l'immediato utilizzo clinico.
Geni terapeutici
Terapia genica delle malattie ereditarie monogeniche

Come accennato, ottenere l'inserimento di DNA esogeno in una sequenza omologa di cellule di mammifero è un evento molto difficile che si verifica con una frequenza di circa l'uno per mille rispetto all'inserimento casuale. Il concetto di terapia genica nasce, quindi, con l'idea di supplire a una funzione cellulare mediante l'introduzione di un gene che codifichi la proteina alterata. Dal momento che, nella maggior parte dei casi, la dimensione dei geni dei Mammiferi è troppo grande per essere manipolata facilmente, l'elemento genico che viene trasferito è il cDNA corrispondente al gene, o, ancor più spesso, solo la sua porzione codificante. La terapia genica delle malattie ereditarie si basa proprio su questa strategia, e cioè sull'aggiunta al genoma della cellula appropriata di un gene che conferisca la funzione mancante a causa delle mutazioni degli alleli endogeni. Come tale, la terapia genica può affrontare soltanto malattie autosomiche o legate al cromosoma X con fenotipo recessivo, e in cui il tessuto appropriato sia accessibile con facilità. L'attuale esclusione delle malattie con eredità dominante, la cui terapia cioè richiede la correzione specifica dell'allele mutato, è legata alla limitatezza delle nostre conoscenze sui meccanismi che regolano la ricombinazione omologa nelle cellule di mammifero. Nella tab. 2 è presentato un elenco di patologie per le quali la sperimentazione di terapia genica è già arrivata in fase clinica.
Terapia genica dei tumori

Gli approcci terapeutici che mirano alla terapia dei tumori si basano sul presupposto che ogni neoplasia derivi, da un lato, dall'alterazione dei meccanismi molecolari che regolano la proliferazione cellulare e, dall'altro, dall'incapacità del sistema immunitario di controllare la proliferazione stessa. I geni terapeutici possono essere classificati in varie categorie. Una prima categoria è costituita dai geni che inducono arresto della proliferazione cellulare, agendo sulle proteine della replicazione del DNA (per es., PCNA, Proliferating cell nuclear antigen), sui meccanismi di controllo del ciclo cellulare e della sua correlazione con il danno genotossico (chinasi cdc-2, p53, p16) o direttamente su oncogeni patologicamente attivati come c-myc e c-fos (tab. 3). Alternativamente, l'aggressione da parte delle cellule del sistema immunitario al tumore può essere resa più efficace mediante trasferimento, nelle cellule del tumore stesso, di geni che codificano per citochine immunoregolatrici (IL-2, IL-12, IL-7, IL-4, GM-CSF e altre). Oppure, ancora, l'immunogenicità stessa del tumore può essere aumentata mediante trasferimento di geni che sono in grado di esprimere antigeni di istocompatibilità diversi da quelli del paziente, o antigeni tumore-specifici. Nel caso delle malattie linfoproliferative delle cellule B, l'antigene specifico che può essere utilizzato a questo scopo è l'anticorpo che viene espresso dal clone neoplastico.
Un approccio alternativo di terapia genica delle malattie neoplastiche è rappresentato dal trasferimento di geni che causano la morte cellulare in maniera controllabile con l'utilizzo di farmaci. Un esempio di questa strategia è l'iniezione intratumorale di vettori virali esprimenti il gene della timidina-chinasi di HSV-1. L'espressione intratumorale di questo enzima è innocua di per sé, ma diventa tossica quando il paziente è trattato con il farmaco ganciclovir, in quanto l'enzima (a differenza della timidina-chinasi delle cellule umane) è in grado di fosforilare il profarmaco e renderlo attivo. Il risultato di questa attivazione è il blocco della sintesi del DNA e la conseguente morte cellulare. Questo tipo di approccio è potenzialmente molto interessante in quanto le cellule trasdotte con il gene della timidina-chinasi, in presenza di ganciclovir, sono in grado di rilasciare metaboliti tossici anche alle cellule immediatamente vicine che non esprimono il gene, creando quindi un effetto di diffusione dell'efficacia terapeutica che va al di là dell'efficienza stessa di trasferimento genico (bystander effect).
Vi è una differenza concettuale importante tra gli approcci che causano il blocco della proliferazione cellulare o inducono una tossicità sito-specifica nel tumore e quelli che mirano ad aumentare l'efficienza di riconoscimento del tumore da parte del sistema immunitario. I primi, infatti, richiedono che tutte le cellule neoplastiche siano direttamente o indirettamente interessate dal trattamento; i secondi, invece, confidano nell'efficacia del sistema immunitario attivato in modo specifico per la distruzione delle cellule, non raggiunte direttamente dal trattamento di terapia genica, e delle metastasi.
Infine, un tipo di approccio alternativo perseguito dalla terapia genica dei tumori consiste nel trasferire geni che consentano di elevare l'indice terapeutico della chemioterapia antitumorale. In molti casi, è la tossicità a livello delle cellule ematopoietiche del midollo osseo a costituire uno dei fattori limitanti nella formulazione della dose del trattamento chemioterapico. Ne consegue che l'induzione di una resistenza in queste cellule, mediante trasferimento genico nella popolazione staminale CD34+, può consentire di elevare la soglia di tollerabilità alla chemioterapia. Tra i geni potenzialmente utilizzabili a questo scopo vi sono quelli della famiglia ABC, che codificano per delle proteine trasportatrici di membrana in grado di prevenire l'accumulo intracellulare di una serie di farmaci antitumorali quali gli alcaloidi della vinca (vincristina, vinblastina), le antracicline (adriamicina, daunorubicina), l'etoposide e il paclitaxel.
Vaccinazioni
Come già accennato in precedenza, uno sviluppo molto importante della terapia genica è rappresentato dalla possibilità di utilizzare questa tecnologia come metodo di vaccinazione. In questo caso, il gene che codifica la proteina contro la quale si voglia provocare una risposta del sistema immunitario può essere espresso in modo transitorio da parte di un vettore virale che non si integra stabilmente nel genoma (per es., adenovirus, poxvirus) o somministrato come DNA nudo, solitamente mediante iniezione intramuscolare o intradermica. Vi sono importanti vantaggi in questo tipo di approccio rispetto alle comuni tecniche vaccinali basate sulla somministrazione di preparazioni contenenti l'agente infettivo inattivato o alcune proteine ricombinanti: tra i principali, la sicurezza della preparazione, il costo ridotto (non c'è bisogno di complesse tecniche di purificazione), la facilità di conservazione e, spesso, la migliore stimolazione del sistema immunitario, poiché la proteina esogena è espressa direttamente dalla cellula del paziente nel contesto dei suoi antigeni di istocompatibilità. Esempi di patologie affrontate attualmente con questo tipo di approccio sono la tubercolosi, la sindrome da immunodeficienza acquisita (AIDS, Acquired immunodeficiency syndrome) e alcuni tipi di tumori (utilizzando antigeni specifici del tumore).
DNA ed RNA non codificanti
Al fine di ottenere un effetto terapeutico mediante introduzione di materiale genetico esogeno, gli studi attuali non sono limitati all'uso di geni che codificano una proteina normale. Nuovi approcci sperimentali mirano all'inattivazione di un RNA messaggero 'patologico' utilizzando i ribozimi, ossia piccole molecole di RNA che si appaiano in maniera specifica a un RNA complementare e sono in grado di catalizzare il taglio enzimatico di quest'ultimo. La specificità di riconoscimento di questi enzimi a RNA può essere modificata per farli reagire con RNA patologici specifici. Questo tipo di approccio appare promettente per la terapia genica delle malattie ereditarie autosomiche dominanti (in cui è richiesto che l'RNA messaggero dell'allele patologico sia distrutto), per le malattie virali (in cui si ricerca la distruzione degli mRNA virali) o per le malattie neoplastiche in cui vi sia la formazione di mRNA cellulari patologici (per es., nella leucemia mieloide cronica, in cui la traslocazione reciproca tra il cromosoma 9 e il cromosoma 22 porta alla formazione del gene di fusione bcr-abl, la cui espressione è responsabile del fenotipo trasformato).
Un approccio concettualmente analogo a quello dei ribozimi è basato sull'inibizione di un mRNA patologico mediante oligonucleotidi a DNA o RNA antisenso, cioè con sequenza nucleotidica complementare a quella del trascritto: l'appaiamento specifico porta, da un lato, all'inattivazione dell'RNA e, dall'altro, alla sua degradazione da parte degli enzimi cellulari. Mentre i ribozimi sono usualmente espressi nel contesto di vettori virali, e quindi prodotti all'interno della cellula trasdotta, gli oligonucleotidi antisenso vengono somministrati direttamente alla cellula dall'esterno, sfruttando la capacità di diversi tipi cellulari di assumere queste molecole e di trasportarle nel nucleo. Come già discusso in precedenza, l'efficacia del secondo tipo di approccio è limitata dalla farmacocinetica in vivo degli oligonucleotidi, dalla necessità di somministrarli in concentrazioni molto elevate e, in diversi casi, dalla loro scarsa specificità di azione.
Gli approcci basati sui ribozimi e sugli oligonucleotidi antisenso vengono oggi progressivamente abbandonati a favore dell'utilizzo di piccoli RNA a doppio filamento che sfruttano il fenomeno dell'interferenza genica basata sull'RNA (RNAi, RNA interference). Corti duplex di RNA di cui un filamento è complementare a un mRNA della cellula (siRNA, Small interfering RNA) sono in grado di innescare un processo enzimatico intracellulare che porta alla degradazione l'mRNA cellulare bersaglio e, quindi, alla diminuzione del livello della relativa proteina.
Strategie di somministrazione dei geni terapeutici
Da quanto esposto fin qui appare evidente che lo sviluppo della terapia genica richiede di considerare diversi aspetti: l'individuazione della patologia che possa trarne vantaggio, la scelta del gene terapeutico, la scelta dei segnali regolativi da associare a esso, lo sviluppo di un sistema di trasferimento genico adeguato, la scelta della via di somministrazione e della posologia adeguate, lo sviluppo di metodiche per valutare il successo del trattamento. Per quanto riguarda le malattie ereditarie, la terapia genica si rivolge, per ora, soltanto a quelle con ereditarietà recessiva o legata al cromosoma X. Inoltre, tutte le cellule del tessuto da trattare devono essere uniformemente accessibili alla terapia, direttamente in vivo o ex vivo. Questo tipo di considerazioni rende problematica la progettazione di un sistema di terapia genica per le malattie ereditarie con compromissione neurologica o che interessino tessuti come quello osseo o muscolare, mentre la indirizzano verso patologie organo-specifiche (fegato, sistema ematopoietico, sistema linfoide, epitelio respiratorio). Un problema particolare è utilizzare la terapia genica per patologie in cui è presente un difetto di proteine secrete, come per esempio nei difetti genetici delle proteine della coagulazione. In questo caso, può risultare conveniente far produrre queste proteine al muscolo o ai fibroblasti impiantati nel derma piuttosto che al fegato. Alternativamente, nel derma del paziente è possibile impiantare dei neo-organi, immunologicamente separati, in cui proliferino cellule eterologhe ingegnerizzate per secernere e immettere nel torrente circolatorio grandi quantità della proteina carente.
Il problema di più difficile soluzione è la corretta scelta del binomio gene terapeutico-sistema di trasferimento genico. Infatti, a seconda del tipo di applicazione, appare critico ai fini del risultato che il trasferimento del gene sia conseguito, nel tessuto bersaglio, in un numero sufficiente di cellule rilevanti. Per la terapia genica delle malattie ereditarie sono tendenzialmente presi in considerazione i vettori retrovirali o adenoassociati per la loro proprietà di trasferire in modo permanente il gene terapeutico nel genoma dell'ospite. Tuttavia, un problema critico a questo proposito è la corretta identificazione della cellula staminale del tessuto che si intende trattare. Come identificare e raggiungere la cellula staminale dell'epitelio respiratorio per trasferire permanentemente il gene della fibrosi cistica? Quale sottotipo di cellule che esprime il marcatore CD34+ è veramente il precursore staminale ematopoietico? Un problema molto affrontato nei laboratori che si occupano di terapia genica con i vettori retrovirali è quello di ottenere livelli di espressione del gene terapeutico elevati, persistenti e, possibilmente, tessuto-specifici. Infatti, in particolare nella correzione delle malattie genetiche, è indispensabile che l'espressione del gene terapeutico si verifichi in maniera il più possibile simile a quella fisiologica.
Un esempio di questo requisito è rappresentato dalla terapia genica delle talassemie, in cui la sintesi delle catene globiniche deve avvenire nell'eritroblasto in maniera bilanciata. Dal momento che le sequenze dei promotori che controllano l'espressione dei geni endogeni non sono, molto spesso, conosciute completamente o sono troppo estese per essere clonate all'interno dei vettori, risulta importante identificare promotori tessuto-specifici abbastanza corti da essere utilizzabili e sufficientemente potenti. Un altro esempio che sottolinea questo concetto viene dalle prime esperienze di terapia genica di malattie che richiedono l'espressione dei geni nelle cellule della serie mieloide. L'espressione della maggior parte dei vettori retrovirali che si integra nei precursori staminali CD34+ tende a essere progressivamente spenta nel corso del differenziamento mieloide, a meno che essa non sia controllata direttamente da promotori fisiologicamente attivi nel granulocita maturo e nel macrofago.
Ovviamente diversa è la situazione per quanto riguarda la terapia genica dei tumori. Nel caso ci si indirizzi al trasferimento nelle cellule tumorali di geni che inducano una risposta immunitaria vigorosa, non è rilevante che tutte le cellule tumorali siano raggiunte dal trattamento, né che il trasferimento genico sia permanente. In questo caso, sono molto appropriati i vettori adenovirali, in grado di esprimere grandi quantità di proteina immunogenica o immunostimolatrice. Gli approcci che si possono utilizzare sono quelli della somministrazione diretta del vettore virale nel tumore mediante iniezione intratumorale, o il trasferimento genico nelle cellule del tumore coltivate in vitro (o in linee cellulari tumorali allogeniche irradiate), seguito dalla loro somministrazione al paziente a scopo vaccinale.
Sperimentazione clinica
Tra il 1986 e il 1987 una prima sperimentazione clinica di trasferimento genico fu praticata negli Stati Uniti, in modo surrettizio, su un bambino talassemico, senza alcun effetto terapeutico. Il comportamento degli sperimentatori fu criticato aspramente dal mondo scientifico e medico e, da allora, in tutti i Paesi industrializzati, ogni sperimentazione clinica di terapia genica deve essere vagliata ed eventualmente approvata da appositi comitati etici locali e nazionali. La prima applicazione di terapia genica all'uomo in questo regime fu approvata negli Stati Uniti dal Recombinant DNA Advisory Committee nell'ottobre del 1988, e la sperimentazione clinica iniziò nei primi mesi del 1989. Si trattava di una sperimentazione di marcatura genica effettuata su dieci pazienti con melanoma, per comprendere se i linfociti isolati dal tumore (TIL, Tumor infiltrating lymphocytes) potessero essere espansi in vitro conservando la capacità di infiltrare il tumore una volta reinfusi. Al fine di distinguerli, i linfociti furono marcati mediante infezione con un vettore retrovirale. Linfociti contenenti il provirus furono trovati nel sangue periferico tre settimane dopo l'infusione in tutti i pazienti e nelle biopsie tumorali per almeno due mesi, dimostrando così sia l'efficacia del trasferimento genico ex vivo sia le proprietà da parte dei TIL di indirizzarsi sul tumore.
Da quel momento si è assistito a un aumento esponenziale delle sperimentazioni cliniche effettuate negli anni successivi. Verso la fine del 1999 si è raggiunto un picco, legato in particolare alla grande attività effettuata negli Stati Uniti, dove si stima che nella seconda metà del 1999 siano stati reclutati per esperimenti di trasferimento genico circa dieci pazienti ogni settimana. La crescente percezione della generale inefficacia dei protocolli proposti, seguita all'iniziale entusiasmo, ha portato a una progressiva riduzione delle nuove sperimentazioni cliniche negli anni successivi, a favore di maggiori sforzi nel miglioramento delle tecnologie di base e del livello delle conoscenze biologiche. A tutt'oggi (2006), sono state completate o sono tuttora aperte più di un migliaio di sperimentazioni. Circa il 60% di queste sperimentazioni è di fase I: esse si propongono cioè di valutare la sicurezza della procedura, con minor attenzione al beneficio terapeutico; tali sperimentazioni sono condotte su un numero limitato di pazienti. Soltanto un terzo delle sperimentazioni è di fase I/II o II, e si propone di valutare, in maniera limitata e preliminare, l'efficacia della terapia oltre che la sua sicurezza. Meno di trenta sperimentazioni ha raggiunto la fase III (valutazione dell'efficacia terapeutica in un grande numero di pazienti). Il limitato numero di sperimentazioni in fase più avanzata della fase I è indicativo dei problemi che la terapia genica sta incontrando a livello clinico. Innanzitutto, i risultati di diversi studi di fase I non si sono rivelati conformi alle aspettative e non hanno dato seguito a ulteriori studi clinici. Basti citare, a questo proposito, le trentaquattro sperimentazioni cliniche riguardanti la terapia genica della fibrosi cistica utilizzando il gene CFTR: sebbene più di metà di queste abbia avuto inizio prima della fine del 1995, nessuna ha raggiunto a tutt'oggi la fase III, in quanto i risultati ottenuti non sono stati incoraggianti.
Patologie affrontate e principali risultati ottenuti
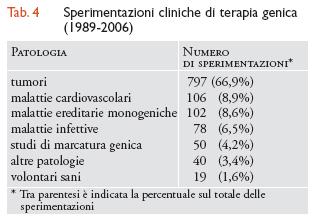
La tab. 4 presenta le sperimentazioni cliniche condotte finora negli Stati Uniti e negli altri Paesi, suddivise per tipo di patologia.
Terapia genica dei tumori. - La maggior parte delle sperimentazioni terapeutiche riguarda la terapia genica dei tumori. Gli approcci terapeutici sono molto diversi e utilizzano, sia in vivo che ex vivo, il trasferimento di geni che causano la soppressione della crescita cellulare, che codificano le citochine o gli antigeni stimolatori del sistema immunitario, secondo i principi delineati in precedenza (tab. 3). Queste sperimentazioni sono dirette principalmente alla terapia di carcinomi o melanomi in fase avanzata (spesso metastatici) in cui il trasferimento genico viene fatto in vivo in sede intratumorale o ex vivo utilizzando cellule neoplastiche autologhe a fini vaccinali. I risultati iniziali di diversi studi hanno dimostrato una marcata riduzione della massa tumorale in seguito a iniezione intratumorale diretta, mediante liposomi, dei geni in grado di stimolare la reattività del sistema immunitario (quali i geni che codificano HLA-B7 e la β2-microglobulina) nei melanomi metastatici e nelle metastasi di tumori del colon. Analogamente, il trattamento con geni che esprimono citochine, di cellule tumorali o di cellule xenogeniche esprimenti gli stessi antigeni, fa generalmente osservare una riduzione della massa tumorale primitiva e talvolta anche delle metastasi. In entrambi questi casi, tuttavia, sembra ancora prematuro azzardare un'ipotesi sulla reale efficacia a medio e lungo termine di queste procedure. In un caso aneddotico un paziente con melanoma metastatico trattato con un retrovirus che esprimeva il gene codificante l'interferone γ, ha visto una completa remissione sistemica della malattia con assenza di tumore per almeno sette mesi dopo il trattamento. Il successo in un singolo paziente, ovviamente, non implica la generale utilità di questo tipo di trattamento.
Nell'ambito della terapia genica dei tumori, un'applicazione particolare è quella utilizzata per le neoplasie cerebrali (glioblastomi, astrocitomi) in cui viene trasferito in sede tumorale con iniezione stereotassica il gene della timidina-chinasi di HSV-1, cui fa seguito il trattamento del paziente con ganciclovir. È stato dimostrato che questo tipo di terapia è in grado di far regredire in maniera significativa i tumori sperimentali nell'animale. Sulla base di questo risultato, è stato condotto un ampio studio di fase III, che ha incluso più di quaranta centri nel Nord America e in Europa, e ha reclutato più di duecento pazienti in cui sono state direttamente iniettate, in corso di intervento chirurgico per la rimozione della massa tumorale, le cellule packaging che producevano un vettore retrovirale in grado di trasdurre la timidina-chinasi. I risultati di questo studio sono stati però deludenti in termini di allungamento della vita dei pazienti.
Risultati potenzialmente interessanti sono stati ottenuti mediante il trattamento con linfociti T specifici per antigeni tumorali, che vengono espansi in vitro e trasdotti con il gene della timidina-chinasi (che potrà poi permettere l'eliminazione selettiva dei linfociti iniettati) nei trapianti di midollo allogenico. In questa sperimentazione, i linfociti T trapiantati svolgono un ruolo preminente, in quanto hanno, da un lato, un effetto positivo nel mediare il riconoscimento delle cellule tumorali (GvL, Graft versus leukemia) e, dall'altro, un effetto negativo, in quanto mediano il danno citotossico anche contro i tessuti normali del ricevente (GvHD, Graft versus host disease, malattia del trapianto contro l'ospite). Otto pazienti ricaduti nella malattia o che avevano sviluppato un linfoma indotto dal virus di Epstein-Barr (EBV) dopo trapianto di midollo allogenico depleto di cellule T, sono stati trattati con linfociti del donatore trasdotti con il gene della timidina-chinasi. I linfociti trasdotti sono sopravvissuti fino a un anno e hanno esercitato attività antitumorale in cinque pazienti. Tre pazienti hanno sviluppato la GvHD, che è stata controllata efficacemente mediante trattamento con ganciclovir. Anche se l'efficacia di questa strategia necessita di essere verificata in un numero più vasto di casi, i risultati preliminari ottenuti sono di indubbio interesse. Infine, alcuni studi americani ed europei hanno utilizzato il gene mdr-1 (un membro della famiglia dei trasportatori ABC) per conferire alle cellule CD34+ trattate ex vivo un'accresciuta resistenza alla terapia antiblastica. Queste cellule sono poi reinfuse nel paziente mediante trapianto autologo.
Terapia genica delle malattie ereditarie monogeniche. - La prima sperimentazione di terapia genica delle malattie ereditarie autorizzata nell'uomo è stata condotta per curare la deficienza di adenosina-deamminasi (ADA), una malattia autosomica recessiva molto rara, ma in teoria molto adatta alla terapia genica, in quanto le cellule corrette hanno un naturale, ancorché modesto, vantaggio proliferativo. La sperimentazione clinica è avvenuta negli Stati Uniti trasferendo nei linfociti T del sangue periferico di due bambini, mediante un vettore retrovirale, il gene che codifica l'enzima. I risultati di questa sperimentazione (cellule geneticamente modificate sono risultate persistere alcuni anni dopo il trattamento) hanno incoraggiato anche altre sperimentazioni praticate sui linfociti o sui precursori ematopoietici CD34+ dell'adulto o del cordone ombelicale. Più di settanta sperimentazioni cliniche sono state eseguite trasducendo le cellule staminali ematopoietiche del midollo osseo ex vivo dopo averne stimolato la proliferazione utilizzando dei cocktail di fattori di crescita prima della somministrazione del vettore retrovirale. L'infezione ex vivo di queste cellule è straordinariamente efficiente. Tuttavia, la loro reinfusione nel paziente non porta alla persistenza di popolazioni cellulari trasdotte, in quanto la maggior parte delle cellule reinfuse perde le proprie potenzialità staminali nel periodo di coltura. Salvo eccezioni, soltanto una piccola percentuale (0,01÷1%) di cellule contenenti il provirus è riscontrabile nel paziente a distanza di anni dal trapianto.
Una ulteriore essenziale limitazione dei vettori retrovirali è legata al progressivo spegnimento dell'espressione del gene veicolato da parte della cellula trasdotta. Questo evento è la conseguenza della metilazione delle citosine nella regione del promotore LTR del vettore, un evento che è associato al rimodellamento della cromatina verso uno stato compattato e non accessibile all'apparato trascrizionale. Questa risposta cellulare all'evento di trasduzione rappresenta probabilmente un meccanismo sviluppatosi evolutivamente per preservare l'integrità dell'espressione dell'informazione genetica della cellula nel confronto dell'integrazione di elementi trasponibili. La perdita della staminalità delle cellule trasdotte e lo spegnimento del gene terapeutico sono stati i principali motivi dello scarso successo della maggior parte delle sperimentazioni finora condotte con questo tipo di approccio. Nel caso stesso della deficienza di ADA, il successo terapeutico è stato raggiunto soltanto quando le cellule corrette ex vivo sono state reinoculate dopo il trattamento dei pazienti con un regime di chemioterapia in grado di rimuovere parte delle cellule del midollo, in modo da fare spazio all'attecchimento delle cellule geneticamente corrette. Un analogo approccio viene ora utilizzato nei protocolli per altre malattie genetiche in cui sia richiesto il trasferimento genico nei precursori ematopoietici, e che comprendono la forma autosomica e quella legata al cromosoma X della malattia granulomatosa cronica, l'anemia di Fanconi, e alcune mucopolisaccaridosi.
Una straordinaria eccezione a queste considerazioni è rappresentata dalla sperimentazione di terapia genica per il difetto della catena γ di alcuni recettori per le interleuchine, difetto responsabile della sindrome da immunodeficienza acquisita legata al cromosoma X (X-SCID). In questa patologia, il trasferimento del gene che codifica per la catena γ nelle cellule CD34+ di midollo ha ormai consentito la guarigione di un numero rilevante di pazienti, dal momento che la correzione del difetto genetico anche in un numero molto esiguo di precursori ematopoietici conferisce loro un notevolissimo vantaggio selettivo, e ne garantisce quindi l'espansione previlegiata e la conseguente ripopolazione dell'intero midollo nell'arco di pochi mesi. La terapia della X-SCID, quindi, rappresenta l'unico vero successo della terapia genica a tutt'oggi. Tuttavia, questa sperimentazione ha portato drammaticamente alla ribalta il problema della mutagenesi inserzionale causata dai vettori retrovirali, in quanto due dei bambini trattati ‒ e apparentemente guariti dalla loro malattia ‒ hanno successivamente sviluppato una leucemia causata dall'integrazione del genoma virale in prossimità dell'oncogene cellulare LMO2. Questa evenienza sta stimolando la comunità scientifica a sviluppare delle strategie per ottenere l'inserzione sito-specifica del DNA trasferito.
Attualmente, circa un terzo delle sperimentazioni cliniche di terapia genica per le malattie monogeniche ha come obiettivo la cura della fibrosi cistica. Tali sperimentazioni tentano di trasferire il gene che codifica la proteina CFTR nell'epitelio respiratorio di pazienti tramite vettori adenovirali di seconda e terza generazione, AAV o liposomi. La somministrazione del virus ricombinante avviene direttamente in vivo, mediante instillazione tramite broncoscopio a diversi livelli dell'albero respiratorio, o mediante aerosol. Nella maggior parte dei casi si tratta di sperimentazioni di fase I, effettuate anche nell'epitelio nasale dei pazienti. In generale, i risultati hanno dimostrato che il gene CFTR può essere trasferito nelle cellule epiteliali delle vie respiratorie e che la sua espressione è in grado di provocare una correzione parziale o quasi totale delle anomalie di voltaggio transepiteliale e di trasporto ionico proprie della malattia. In generale, questa correzione è però transitoria e dura solo un paio di settimane dopo il trattamento. Per quanto riguarda i vettori, questi studi clinici hanno confermato l'efficacia degli adenovirus in termini di efficienza di trasferimento genico, ma hanno anche portato chiaramente alla luce la loro principale limitazione, ovvero la proprietà di indurre una forte risposta infiammatoria e immunitaria da parte dell'ospite. Questa risposta limita in maniera significativa il raggiungimento dell'obiettivo terapeutico, previene la somministrazione ripetuta di vettore e, soprattutto, induce una situazione patologica di potenziale gravità. La potenziale pericolosità dei vettori adenovirali è venuta alla luce inizialmente nel corso di una sperimentazione per la terapia genica della fibrosi cistica e in seguito, nel contesto di una sperimentazione per la terapia di un'altra malattia ereditaria monogenica, il deficit di ornitina-transcarbammilasi (OTC), un enzima epatico del ciclo dell'urea. Nel 1989, un paziente con deficit di OTC è morto dopo l'inoculazione di un vettore adenovirale di prima generazione nel fegato. Questo evento ha drammaticamente sollevato l'esigenza di sviluppare vettori adenovirali di generazione avanzata, in cui i geni proinfiammatori del virus siano rimossi.
Infine, vanno menzionate due applicazioni recenti della terapia genica, entrambe ottenute utilizzando vettori AAV. La prima si è rivolta alla cura dell'emofilia B e ha visto condurre due sperimentazioni nelle quali il gene per il fattore IX è stato veicolato da questi vettori nel muscolo o nel fegato dei pazienti. La seconda è appena iniziata e si rivolge alla terapia genica di una forma di cecità congenita (l'amaurosi congenita di Leber), in cui il gene per la proteina RPE65 viene veicolato da AAV nella retina dei bambini affetti. In entrambi i casi, analoghi approcci di terapia genica con vettori AAV hanno avuto pieno successo terapeutico nella sperimentazione in modelli animali di queste malattie.
Terapia genica dell'AIDS. - Verso la metà degli anni Novanta, molte sperimentazioni cliniche si sono rivolte alla terapia genica dell'infezione da HIV. Alcune erano basate sull'utilizzo di vettori per la vaccinazione genetica contro alcune proteine del virus, nel tentativo di potenziare la risposta immunitaria dei pazienti infettati. Altre sperimentazioni si proponevano di trasferire linfociti citotossici CD8+ autologhi o allogenici, stimolati in vitro contro antigeni virali, in modo da trasferire in maniera adottiva una risposta vigorosa contro il virus. Infine, un altro approccio prevedeva l'utilizzo di geni terapeutici in grado di rendere la cellula trasdotta resistente all'infezione. Questi geni sono rappresentati da ribozimi in grado di tagliare il genoma virale, dal mutante transdominante negativo M10 della proteina virale Rev, o da un gene codificante un anticorpo intracellulare contro la proteina Env. Molte di queste sperimentazioni sono state interrotte, oppure il loro risultato non è stato facilmente valutabile, a causa dell'introduzione della moderna terapia di combinazione per l'infezione da HIV-1, che prevede l'assunzione di associazioni di farmaci contro molteplici bersagli del virus. Nella maggior parte dei pazienti, questa terapia, pur non essendo in grado di eradicare la malattia, riduce la carica virale a livelli non dosabili, e rende quindi problematica la valutazione dell'efficacia di un eventuale trattamento con terapia genica. Negli ultimi anni, la progressiva insorgenza di ceppi virali mutati e resistenti ai farmaci, e la comparsa nei pazienti di importanti effetti collaterali che impediscono una stretta aderenza alla terapia, stimolano l'allestimento di nuove sperimentazioni con l'obiettivo di rendere le cellule CD4+ resistenti all'infezione virale.
Terapia genica delle malattie cardiovascolari. - Hanno recentemente assunto grande rilevanza nel panorama di applicazioni della terapia genica quelle rivolte alle malattie cardiovascolari, sia per la prevalenza e l'importanza economica e sociale di queste patologie nella popolazione occidentale, sia per lo sviluppo, da parte della cardiologia interventistica, di tecnologie di rilascio dei vettori per la terapia genica direttamente nei distretti vascolari in cui l'applicazione è richiesta. Il principale obiettivo di queste sperimentazioni è quello di trasferire nei tessuti ischemici geni in grado di stimolare il processo di neoangiogenesi. I principali di questi geni sono quelli delle famiglie del Vascular endothelial growth factor (VEGF) e del Fibroblast growth factor (FGF), che agiscono stimolando l'attivazione e la proliferazione delle cellule endoteliali e la conseguente formazione di un nuovo albero vascolare. I pazienti cui questa terapia è rivolta soffrono di malattia ischemica vascolare degli arti o cardiaca. Le sperimentazioni iniziali sono state condotte mediante trasferimento genico con DNA plasmidico; più recentemente, sono state ampliate con una serie di sperimentazioni con vettori adenovirali di prima generazione.
Studi di marcatura genica. - Un numero significativo di sperimentazioni cliniche finora condotte non ha riguardato propriamente la terapia genica, in quanto non mirava a un beneficio terapeutico del paziente, bensì aveva lo scopo di ottenere informazioni biologiche e cliniche importanti tramite studi di marcatura genica. In generale, si trasferisce un gene marcatore, inserito in un vettore virale, in determinate popolazioni cellulari ex vivo, al fine di studiare il destino di queste popolazioni dopo re-introduzione nel paziente. Molti di questi studi sono indirizzati a risolvere problematiche di ematologia sperimentale, quali, per esempio, la valutazione comparativa dell'efficienza di diverse procedure per la mobilizzazione, la purificazione e la manipolazione delle cellule staminali ematopoietiche ottenute dal midollo osseo o dal sangue periferico. Gli studi di marcatura genica hanno anche permesso di stabilire in maniera inequivocabile che l'origine delle ricadute di tumori ematologici o solidi in pazienti sottoposti a trapianto di midollo autologo deriva dalla presenza di cellule tumorali nel trapianto, e non alla persistenza di cellule tumorali nel paziente. In questa linea, gli studi di marcatura delle cellule di midollo consentono anche di paragonare l'efficienza di diverse metodiche di purging (purificazione) nel trapianto autologo. Un altro campo di utilizzo di questo tipo di sperimentazioni di marcatura genica è lo studio delle cinetiche delle popolazioni di linfociti con potenziale attività antitumorale, e cioè i linfociti che infiltrano i tumori o i linfociti con funzione citotossica dopo attivazione con citochine, le cellule LAK (Lymphokin-activated killer), ovvero cellule killer attivate da linfochine.
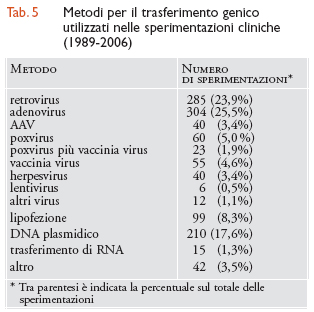
Metodologie di trasferimento genico utilizzate nella sperimentazione clinica. - Quasi due terzi delle sperimentazioni condotte finora si sono avvalse di vettori virali, a causa della loro efficienza nel veicolare i propri geni all'interno della cellula infettata. I vettori basati sui retrovirus, che costituivano il principale metodo di trasferimento genico all'inizio degli anni Novanta, sono stati progressivamente abbandonati per la loro incapacità di trasdurre cellule quiescenti e per la caratteristica di essere sottoposti a silenziamento dell'espressione genica nel tempo. Rimangono utilizzati prevalentemente per le sperimentazioni ex vivo che prevedono il trasferimento genico nei precursori ematopoietici. I vettori basati su adenovirus di prima generazione sono utilizzati in circa un quarto delle sperimentazioni, ma continuano a suscitare problematiche legate alla loro proprietà di stimolare una forte risposta immunitaria e immunogenica. Assai promettenti sono invece i vettori basati su AAV, il cui uso si sta progressivamente estendendo nelle sperimentazioni che prevedono il trasferimento genico nel muscolo, nel cuore, nel cervello e nella retina. Formulazioni basate sui liposomi e su complessi DNA/lipidi cationici/proteine sono utilizzate in meno del 10% delle sperimentazioni, a causa della loro relativa inefficacia quando comparate all'utilizzo di vettori virali (tab. 5).
Considerazioni etiche
Ancor prima che iniziasse una vera e propria sperimentazione clinica della terapia genica, la comunità scientifica ha dibattuto ampiamente sulla liceità d'intervento modificativo sul genoma umano, giungendo ad alcune conclusioni accettate pressoché universalmente e, in alcuni Paesi, anche codificate in via legislativa. Queste possono essere così riassunte: per quanto riguarda la terapia genica delle cellule somatiche ‒ un intervento che non modifica il patrimonio genetico trasmissibile alla progenie dell'individuo trattato ‒ non vi sono controindicazioni etiche o deontologiche, se non quelle che riguardano tutti i tipi di sperimentazione clinica, vale a dire, essenzialmente, una valutazione seria e prudente del rapporto tra rischi e benefici. Per quanto concerne invece l'intervento sulla linea germinale al fine di guarire una grave malattia ereditaria, intervento che, attualmente, è di sicuro tecnicamente realizzabile in modo analogo a quanto avviene per gli altri Mammiferi, esso viene considerato illecito, almeno allo stato presente delle tecnologie. Infatti, come indicato nei paragrafi precedenti, l'introduzione di materiale genetico all'interno del genoma di cellule di mammifero avviene quasi sempre in modo casuale, e solo molto raramente un gene introdotto dall'esterno va a ricombinarsi con l'eventuale sequenza omologa presente nel cromosoma ricevente. Nella terapia genica somatica si può ovviare a questa limitazione, pur grave, selezionando la cellula in cui si è verificato l'evento ricombinativo più efficace al fine terapeutico, lasciando che la selezione elimini tutte le altre cellule. Questa considerazione ci fa immediatamente capire perché il rapporto costi/benefici di un simile intervento sulla linea germinale non possa essere accettabile allo stato attuale, in quanto comporterebbe necessariamente l'eliminazione di molti prodotti del concepimento nei quali non sia avvenuto l'evento di ricombinazione desiderato: una situazione quindi peggiore di quanto si possa ottenere per lo stesso fine mediante la diagnosi prenatale e l'eventuale interruzione della gravidanza.
Più recentemente, negli Stati Uniti, alcuni biologi, e principalmente James Watson, hanno sostenuto che, in vista del possibile superamento delle difficoltà tecniche, si dovrebbe eliminare qualsiasi remora legislativa alla terapia genica germinale. Anche a questa presa di posizione si è ragionevolmente obiettato che, pure nel caso in cui si potessero realmente risolvere quei problemi tecnici, non si vedono ovvi vantaggi della terapia genica germinale rispetto alla cosiddetta 'diagnosi preimpianto', ossia alle procedure che prevedono la fecondazione in vitro, lo sviluppo dell'uovo fecondato fino allo stadio di due o quattro cellule, l'analisi delle singole cellule per determinare se l'eventuale mutazione dannosa sia presente e il reimpianto delle cellule senza mutazione. Appare pertanto probabile che il bando della terapia genica germinale (legale o autoimposto dalla deontologia) sia destinato a permanere.
Dal momento che l'utilizzo della terapia genica somatica per il trattamento di malattie gravi e inguaribili è attualmente accettato come appropriato dal punto di vista etico, e considerando che gli effetti dannosi della terapia genica appaiono controllabili, il rischio paradossale è che si possa ricorrere alle metodiche di ingegnerizzazione cellulare anche per il trattamento di condizioni non propriamente patologiche, ma cosmetiche. Un esempio di tale problema è stato recentemente sollevato, negli Stati Uniti, dalla richiesta di una società di biotecnologie di utilizzare il trasferimento genico del gene della tirosinasi nelle cellule dei follicoli piliferi per il trattamento della perdita dei capelli che consegue alla chemioterapia nei pazienti con neoplasie. Il possibile passaggio da questo tipo di utilizzo ‒ che, naturalmente, appare ancora eticamente appropriato ‒ all'applicazione della medesima tecnologia per il trattamento della calvizie nelle persone sane appare ovviamente molto breve. Più in generale, questo esempio è illuminante della problematica più estesa riguardante il possibile utilizzo dell'ingegneria genetica nell'uomo per una serie molto vasta di scopi possibili, senza una propria indicazione terapeutica. Il concomitante progresso, da un lato, nella conoscenza dei geni umani e, dall'altro, nel miglioramento delle tecniche di trasferimento genico, rende questo tipo di problematica imminente.
Conclusioni
Da quanto descritto finora risulta evidente che gli entusiasmi inizialmente riposti nella terapia genica devono essere smorzati alla luce dei problemi che sono stati incontrati invariabilmente nei diversi campi di applicazione. Esistono ancora molti traguardi sperimentali che devono essere raggiunti prima che la terapia genica possa rappresentare uno strumento terapeutico propriamente detto. I più importanti possono essere così riassunti: (a) sono necessarie metodologie sicure ed efficienti per ottenere cellule staminali dei principali tessuti bersaglio (sistema emopoietico, epiteli delle mucose, epatociti, ecc.). Questo tipo di studi non è direttamente pertinente alla disciplina della terapia genica, ma rappresenta attualmente un requisito non rinunciabile per assicurarne il successo; (b) è indispensabile sviluppare tecnologie altamente efficienti per trasferire geni all'interno del massimo numero possibile di cellule dell'organo bersaglio e solo, e specificamente, di quello. A questo scopo, è auspicabile lo sviluppo di nuovi vettori che riconoscano e utilizzino recettori specifici di determinati tipi cellulari. Questo fine può essere ottenuto, per esempio, modulando la specificità di riconoscimento della proteina di superficie dei vettori retrovirali, o inserendo nel pericapside retrovirale ligandi che riconoscano specifiche proteine della matrice extracellulare; (c) è necessario limitare la risposta immunitaria contro le cellule ingegnerizzate che producono proteine non presenti nell'organismo ricevente e contro le eventuali proteine prodotte dal vettore per il trasferimento genico. Mentre questo tipo di reattività è utile e viene sfruttato, per esempio, nella terapia genica dei tumori, esso presenta ovviamente degli svantaggi nella terapia genica delle malattie ereditarie; (d) è estremamente utile mettere a punto metodi che conferiscano un vantaggio selettivo alle cellule curate mediante trasferimento genico. Questo approccio consentirà di superare il problema della limitazione nel numero di cellule in cui avviene il trasferimento genico, favorendone l'espansione all'interno dell'organismo ricevente stesso; (e) assumendo che si riesca a ottenere un sistema efficiente e specifico di trasferimento genico, è indispensabile garantire che il gene terapeutico venga espresso in maniera stabile e regolata nella cellula bersaglio. In molte occasioni, l'utilizzo di promotori estranei, ‒ specie se di origine virale, quali il promotore del virus SV40 (Simian virus 40) o di alcuni retrovirus murini o aviari ‒ è svantaggioso, in quanto essi vengono inattivati dalla cellula mediante metilazione o altri meccanismi. Inoltre, molti geni umani importanti, quali quello dell'insulina, non sono espressi in maniera continua, ma rispondono a una serie di segnali fisiologici nell'organismo. Tuttavia, la conoscenza di promotori cellulari specifici per alcuni tessuti od organi è, in molti casi, ancora primitiva.
A volte, invece, gli elementi regolatori della trascrizione comprendono regioni molto vaste di cromatina. Per esempio, le regioni regolatrici dell'espressione della β-globina sono disperse in più di 100 kb, una regione di gran lunga più estesa di quella clonabile in un vettore virale della generazione attuale. Mentre i traguardi elencati si propongono l'indispensabile perfezionamento delle tecnologie attualmente disponibili per la terapia genica, il problema concettualmente più arduo riguarda indubbiamente la messa a punto di metodologie che permettano di integrare il gene esogeno in una sequenza precisa del genoma: in altre parole riuscire a ottenere, con alta efficienza e in maniera esclusiva, eventi di ricombinazione omologa, analogamente a quanto si verifica abitualmente nei batteri, nei lieviti, e anche in determinate linee cellulari aviarie.
La risoluzione di questo problema, ancor prima delle difficoltà di natura tecnologica, richiede tuttavia la comprensione di importanti questioni di carattere generale di biologia molecolare delle cellule dei Mammiferi. Un approccio alternativo alla ricombinazione omologa potrebbe essere fornito dalla realizzazione di veri e propri cromosomi artificiali soprannumerari da introdurre nelle cellule; anche questo progetto richiede la risoluzione di importanti problemi fondamentali, quali la definizione dei fattori che determinano la stabilità di un cromosoma (dimensioni, natura del centromero e dei telomeri) e la sua capacità di replicazione in sincronia con le fasi del ciclo cellulare (identificazione delle origini di replicazione e dei fattori che ne regolano la funzionalità). A dispetto dei problemi delineati più sopra, nella comunità scientifica permane comunque la profonda convinzione che gli approcci di terapia genica siano destinati, in un futuro non lontano, a svolgere un ruolo importantissimo nella pratica medica. Dal momento che non sono permesse scorciatoie o fughe in avanti, sarà indispensabile svolgere un lavoro molto approfondito per comprendere e padroneggiare alcuni processi biologici fondamentali dell'organismo umano.
bibliografia
Aiuti 2002: Aiuti, Alessandro e altri, Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning, "Science", 296, 2002, pp. 2410-2413.
Anderson 1998: Anderson, William F., Human gene therapy, "Nature", 392 (suppl.), 1998, pp. 25-30.
Anson 2006: Anson, Donald S. - Smith, Gregory J. - Parsons, David W., Gene therapy for cystic fibrosis airway disease - is clinical success imminent?, "Current gene therapy", 6, 2006, pp. 161-179.
Arsic 2003: Arsic, Nicola e altri, Induction of functional neovascularization by combined VEGF and angiopoietin-1 gene transfer using AAV vectors, "Molecular therapy", 7, 2003, pp. 450-459.
Barranger 1996: Barranger, John A., Haematopoietic stem cell gene transfer, "Gene therapy", 3, 1996, pp. 379-380.
Baum 1996: Baum, Christopher e altri, Gene transfer to augment the therapeutic index of anticancer chemotherapy, "Gene therapy", 3, 1996, pp. 1-3.
Baum 2003: Baum, Christopher C. - von Kalle, Christof, Gene therapy targeting hematopoietic cells: better not leave it to chance, "Acta haematologica", 110, 2003, pp. 107-109.
Berkner 1992: Berkner, Kathleen L., Expression of heterologous sequences in adenoviral vectors, "Current topics in microbiology and immunology", 158, 1992, pp. 39-66.
Blaese 1995: Blaese, R. Michael e altri, T lymphocyte-directed gene therapy for ADA-SCID: initial trial results after 4 years, "Science", 270, 1995, pp. 475-480.
Bonini 1997: Bonini, Chiara e altri, HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia, "Science", 276, 1997, pp. 1719-1724.
Brenner 1996: Brenner, Malcolm K., Gene marking, "Gene therapy", 3, 1996, pp. 278-279.
Connors 1995: Connors, Thomas A., The choice of prodrugs for gene directed enzyme prodrug therapy of cancer, "Gene therapy", 2, 1995, 702-709.
Culver 1992: Culver, Kenneth W. e altri, In vivo gene transfer with retroviral vector-producer cells for treatment of experimental brain tumors, "Science", 256, 1992, pp. 1550-1552.
Curiel 1991: Curiel, David T. e altri, Adenovirus enhancement of transferrin-polylysine-mediated gene delivery, "Proceedings of the National Academy of Sciences USA", 88, 1991, pp. 8850-8854.
Ferrari 1997: Ferrari, F.K. e altri, New developments in the generation of Ad-free, high-titer rAAV gene therapy vectors, "Nature medicine", 3, 1997, pp. 1295-1297.
Gaspar 2003: Gaspar, H. Bobby - Howe, Steven - Thrasher, Adrian J., Gene therapy progress and prospects: gene therapy for severe combined immunodeficiency, "Gene therapy", 10, 2003, pp. 1999-2004.
Geller 1993: Geller, Alfred I., Herpesviruses: expression of genes in postmitotic brain cells, "Current opinion in genetics and development", 3, 1993, pp. 81-85.
Isner 1996: Isner, Jeffrey M. e altri, Clinical evidence of angiogenesis after arterial gene transfer of phVEGF165 in patient with ischaemic limbo, "Lancet", 348, 1996, pp. 370-374.
Jones, Tuddenham 1995: Jones, Lynelle K. - Tuddenham, Edward G., Gene therapy for the haemophilias, "Gene therapy", 2, 1995, pp. 699-701.
Kay, Hoogerbrugge 1997: Kay, Mark A. - Hoogerbrugge, P.M., Gene therapy, "Proceedings of the National Academy of Sciences USA", 94, 1997, pp. 12.744-12.746.
Ledley 1995: Ledley, Fred D., Nonviral gene therapy: the promise of genes as pharmaceutical products, "Human gene therapy", 6, 1995, pp. 1129-1144.
Matteucci, Wagner 1996: Matteucci, Mark D. - Wagner, Richard W., In pursuit of antisense, "Nature", 384, 1996, pp. 20-22.
Miller 1990: Miller, A.D., Retrovirus packaging cells, "Human gene therapy", 1, 1990, pp. 5-14.
Miller 1992: Miller, A.D., Retroviral vectors, "Current topics in microbiology and immunology", 158, 1992, pp. 1-24.
Miller, Whelan 1997: Miller, Nicholas - Whelan, James, Progress in transcriptionally targeted and regulatable vectors for genetic therapy, "Human gene therapy", 8, 1997, pp. 803-815.
Nabel 1994: Nabel, Gary J. e altri, Immunotherapy for cancer by direct gene transfer into tumors, "Human gene therapy", 5, 1994, pp. 57-77.
Naldini 1996: Naldini, Luigi e altri, In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector, "Science", 272, 1996, pp. 263-267.
Pulkkanen, Yla-Herttuala 2005: Pulkkanen, Kalevi J. - Yla-Herttuala, Seppo, Gene therapy for malignant glioma: current clinical status, "Molecular therapy", 12, 2005, pp. 585-598.
Rosenberg 1990: Rosenberg, Steven A. e altri, Gene transfer into humans-immunotherapy of patients with advanced melanoma, using tumorinfiltrating lymphocytes modified by retroviral gene transduction, "New England journal of medicine", 323, 1990, pp. 570-578.
Ross 1996: Ross, Gail e altri, Gene therapy in the United States: a five-year status report, "Human gene therapy", 7, 1996, pp. 1781-1790.
Rossi 1995: Rossi, John J., Controlled, targeted, intracellular expression of ribozymes: progress and problems, "Trends in biotechnology", 13, 1995, pp. 301-306.
Rubin 1994: Rubin, Joseph e altri, Phase I study of immunotherapy of hepatic metastases of colorectal carcinoma by direct gene transfer, "Human gene therapy", 5, 1994, pp. 1385-1399.
Shi 2003: Shi, Ying, Mammalian RNAi for the masses, "Trends in genetics", 19, 2003, pp. 9-12.
Tinsley, Eriksson 2004: Tinsley, Rogan - Eriksson, Peter, Use of gene therapy in central nervous system repair, "Acta neurologica Scandinavica", 109, 2004, pp. 1-8.
Verma, Somia 1997: Verma, Inder M. - Somia, Nikunj, Gene therapy: promises, problems and prospects, "Nature", 389, 1997, pp. 239-242.
Yang 1990: Yang, Ning-Sun e altri, In vivo and in vitro gene transfer to mammalian somatic cells by particle bombardment, "Proceedings of the National Academy of Sciences USA", 87, 1990, pp. 9568-9572.
Zentilin 1996: Zentilin, Lorena e altri, Functional reconstitution of oxidase activity in X-linked chronic granulomatous disease by retrovirus-mediated gene transfer, "Experimental cell research", 225, 1996, pp. 257-267.